This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The Central Drugs Standard Control Organisation (CDSCO) has requested stakeholders to take appropriate action for timely reporting of adverse events related to medical devices to the Materiovigilance Programme of India (MvPI), in the backdrop of shifting the segment to licensing regime.
The Union ministry of health and family welfare has said that most of the e-pharmacies in the country have informed the Central Drugs Standard Control Organisation (CDSCO) that they are only providing an online platform connecting the users and the licensed pharmacies, in response to the show-cause notice from the drug regulator sent early this […] (..)
With the chemists and druggists across the country opposing the sale of drugs through online, the Central government is taking a stand that the sale of medicines should be strictly under the provisions of the existing regulations and the State Licensing Authorities (SLAs) are legally empowered to act against violation of the legal provisions.
The Delhi High Court has granted ten days’ time to the Government of India and the nation’s drug regulator to file a counter affidavit on the petitions filed by almost 28 pharma companies against the order prohibiting manufacturing, distribution and sale of 14 FDCs licensed prior to the year 1988, in the beginning of June. […]
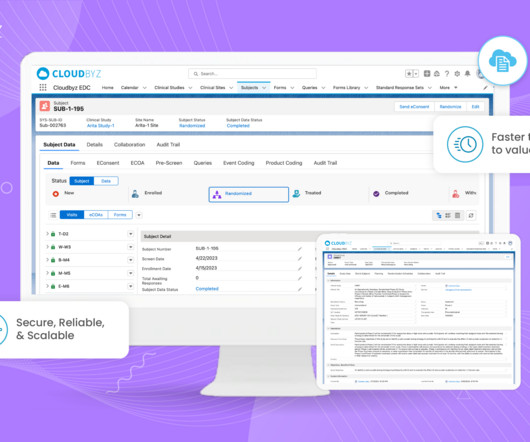
Advantages and Operational Challenges in Adaptive MRCTs Adaptive trial designs are gaining popularity in clinicalresearch due to their ability to improve trial efficiency and uphold ethical standards, resulting in faster and more reliable outcomes.
This prestigious acknowledgment places Cloudbyz among the top-performing companies globally, known for delivering scalable and intuitive CTMS solutions that empower clinicalresearch teams. Global Regulatory Compliance: Ensures compliance with 21 CFR Part 11 and other global regulations, reducing compliance risks.
As few as 2% have peer-reviewed research to back up their claims. ”. These analyses focused exclusively on wellness and smoking cessation apps, bypassing a category of digital therapeutics regulated by the FDA that make scientifically validated claims using sound research. Consistent Standards Matter.
Rebecca Sanders from Lipodystrophy UK tells us how the patient voice helped convince NICE to approve a much-needed drug for this rare disease, and explores how regulators and pharma companies can help make patient involvement in HTA more impactful. This article appears in our free digital magazine Deep Dive: Market Access 2021.
BioPharma Dive interviewed our Christine Moore on how to conduct psychedelic clinicalresearch. With regulators and lawmakers seemingly on board, sponsors have more opportunities than ever to pursue clinicalresearch in this high-velocity area. Psychedelic products are having a renaissance moment. billion in 2027.
Many guidelines focus narrowly on clinical trials that are intended to generate data for regulatory submission to support a new drug license. Many guidelines focus narrowly on clinical trials (including non-randomized studies) that are intended to generate data for regulatory submission to support a new drug license.
PMRs/PMCs are studies or clinical trials that are conducted by the applicant after the Food and Drug Administration (FDA) has approved or licensed a product for marketing. These studies or clinical trials can be required either by regulation or statute (PMR), or by a written agreement, between FDA and the applicant (PMC). .
Immunomedics had licensed the rights to the drug in Asian markets to Everest in 2019 for $65 million upfront and with up to $710 million in potential milestones, including $65 million due on US approval. The new deal means that the licensing deal has been terminated, and the Chinese pharma is no longer on the hook for any remaining payments.
This post covers the key cost drivers for medical device clinical trials. If you are a researcher or financial analyst working in clinicalresearch space or simply curious about clinical trial costs, this post will serve you well. ClinicalResearch Assistants or Associates (CRAs). Patient Grant.
Technological advances, changing patient preferences and evolving guidance from regulators and policy makers have all supported new research methods that break down barriers to participation.
There has been a growing sense of hesitancy about whether regulators will remain equally receptive to these decentralized methods when the pandemic recedes. The Food and Drug Administration (FDA) issued a draft guidance in May 2023 that provided guidance about the conduct of decentralized clinical trials.
Even as a teen in Egypt, Amira Nada was drawn to the idea of clinicalresearch and how it could help people live healthier, richer lives. They weren’t getting the relief they needed, and I wanted to help change that,” Amira adds. She decided to focus on patient recruitment to promote diversity in clinical trial populations.
Compliance and Security: Ensuring compliance with ever-changing regulatory guidelines is a critical concern in clinical trial management. Point systems complicate this task by requiring separate updates and validations to meet new regulations, a process that is both time-consuming and prone to oversights.
Few industries have a need to understand the complexities of compliance more than clinicalresearch. The importance of compliance in clinicalresearch cannot be stressed enough. But the scope of risk extends beyond the tangible to more intangible areas such as organizational reputation and trust.
For example, non-medical personnel should not assess adverse event causality or confirm the clinical significance of lab results. A cross-check ensures curriculum vitae (CVs), and medical licenses (if appropriate) are on file and current. In some countries, local regulations may indicate the sub-investigators need to sign as well.
Avoiding “Customs Chaos” in Clinical Trials Means Working Closely with Your Importer of Record Many global clinical trial shipments get muddled regarding the critical role and responsibilities of the importer of record (IOR) for clinical trials.
Avoiding “Customs Chaos” in Clinical Trials Means Working Closely with Your Importer of Record Many global clinical trial shipments get muddled regarding the critical role and responsibilities of the importer of record (IOR) for clinical trials.
The regulator defines these mission-critical inspections as those that would impact US patients’ access to medical products used to treat, diagnose or prevent serious diseases for which there is no other option available. But drugmakers aren’t the only ones feeling the effects of fewer FDA inspections.
Investigation of the possibility of VAED is crucial during clinical trials for decision-making on vaccine licensing by regulatory agencies. In the majority of cases, the risk of VAED is brought to light during preclinical stages or early clinical trial phases.
XTALKS CLINICAL EDGE: Issue 2 — CSL Seqirus’ Interview Xtalks Clinical Edge is a magazine for clinicalresearch professionals and all who want to be informed about the latest trends and happenings in clinical trials.
PMRs/PMCs are studies or clinical trials that are conducted by the applicant after the Food and Drug Administration (FDA) has approved or licensed a product for marketing. These studies or clinical trials can be required either by regulation or statute (PMR), or by a written agreement, between FDA and the applicant (PMC).
“I am very excited for the field because I feel like we’re beginning to get to a critical mass, where a single method or product can be deemed safe and then adapted for many uses,” said Dr. Peter Marks, head of FDA’s CBER – the organization responsible for regulating gene therapies.
Centralization of Documents When multiple studies share regulatory documents, it is most efficient to centralize them, often done electronically in an eRegulatory management system (eReg).
Proposals from the Committee on the Environment, Public Health and Food Safety (ENVI) address how medicines are regulated in the European Union and, to a lesser extent, overseas. A draft report from the European Parliament health committee aims to ensure access to medicines, promote competitiveness and improve crisis-response mechanisms. .
In rare disease trials, it’s not always feasible to choose clinically-relevant endpoints to measure the efficacy of a new therapeutic. Surrogate endpoints were used as the basis for approval of 45 percent of new drugs reviewed by the FDA between 2010 and 2012.
The NICE recommendation was based on results from the Novartis ORION clinicalresearch program, including Phase III trials ORION-9, ORION-10 and ORION-11, which involved over 3,600 patients and assessed the safety, efficacy and tolerability of inclisiran in lowering LDL-Cholesterol levels 7,8,9.
Data is typically entered into an eCRF by clinicalresearch staff, such as study coordinators or investigators, directly through a secure web-based interface. FDA regulations that establish the criteria for electronic records and electronic signatures. How do eCRFs support the integration of imaging data in clinical trials?
.” This public-spirited industry still required greater oversight, however, and government regulations on medicines increased on both sides of the Atlantic. Thalidomide and the development of drug safety regulation and monitoring. George Merck on the cover of Time magazine.
“Rare disease clinical trials are complex due to the additional scientific, medical, operational and regulatory requirements of newly emerging advanced therapies, such as gene therapy,” says Dr. Terence Eagleton, MB BS, Senior Medical Director at the global clinicalresearch organization (CRO) Medpace.
In clinicalresearch, Cepharanthine exhibits multiple pharmacological properties including anti-oxidative, anti-inflammatory, immuno-regulatory, anti-cancer, anti-viral and anti-parasitic properties. Persons”, as such term is defined in Regulations under the U.S. PharmaDrug Inc.
Our highly experienced clinical team will be placing 2-Bromo-LSD into multi-center United States clinicalresearch locations without restriction.”. “With the acquisition of this intellectual property, we have solved the problem that has plagued psychedelic therapies for so many decades.
Dears Please find hereinafter the latest news related to Clinical Trials in the EU with a respective indirect tax and pharma regulatory licensing impact: 1. The CTIS database is publicly available and leads to increased transparency for indirect tax & pharma regulatory licensing. • What does it mean for you?
wearable device carried by patients to measure certain health-related parameters, remote patient monitoring) and tele-healthcare in clinical trials (e.g. There is a huge opportunity for pharma companies to capitalise on by focusing on increased DEI in clinical trials. video consultations), health data analytics (e.g. What comes next?
Per the agreement between the foundation and Merck, the foundation, in its role as a funder, intends to provide grant funding to the International ClinicalResearch Center (ICRC) at the University of Washington Department of Global Health, which is collaborating with Merck on the IMPOWER 22 study. Merck’s Commitment to HIV.
A risk-benefit approach underpins the decision-making process to evaluate human drugs, drug/device combinations and advanced therapy medicinal products for licensing of human medicines. This is underpinned by historical mistrust in healthcare organisations, governments, and clinicalresearch, which is still prevalent in some communities.
In the pivotal Phase III VERIFY study, researchers evaluated rusfertide, a firstinclass investigational hepcidin mimetic peptide therapeutic designed to mimic the natural hormone that regulates iron, to see if it could reduce the need for regular blood removals (phlebotomies).
based Avacta Group entered into a license agreement with Astrea Bioseparations that allows that company to use Avacta’s Affimer platform in affinity purification applications. Elsewhere around the world: Avacta Group – U.K.-based Shanghai Haini Pharmaceutical — China’s Shanghai Haini Pharmaceutical Co.,
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








































Let's personalize your content