This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The Union health ministry has issued a draft notification framing the rules related to compounding of offences for minor offences under the drug regulations.
Although provisions of the Pharmacy Practice Regulations (PPR) 2015 are not there in the Drugs and Cosmetics (D&C) Act 1940 but are framed under the Pharmacy Act 1948, the steps now being taken by the drugs control administration (DCA) in Tamil Nadu in a motive to strengthen the dispensing activities in pharmacies are completely agreeing […] (..)
Taking into account the four main components of regulator, industry, trade and patient/consumer in a medicine value supply chain, the Drugs and Cosmetics (D&C) Rules, 1945 and D&C Act, 1940 needs an urgent revamp as there is no clause of compensation for ADRs in patients, according to drug regulatory experts.
The Drug Controller General of India (DCGI) has issued a show cause notice to online pharmacies including Tata 1mg for allegedly stocking and selling drugs in contravention of the provisions of the Drugs and Cosmetics Act, 1940 and Rules, once again bringing back the long pending issue of lack of regulations on e-pharmacies under the […]
In this Friday's PCT Grand Rounds, Susan Winckler of the Reagan-Udall Foundation for the FDA will present "Strategies for Improving Public Understanding of FDA and the Products It Regulates: Why Should We Care, and What Might We Do?" The Grand Rounds session will be held on Friday, February 2, 2024, at 1:00 pm eastern.
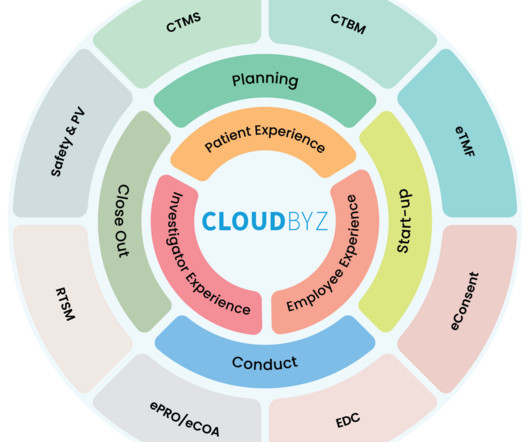
Clinicaltrials are the backbone of drug development, and managing these trials efficiently is paramount. In recent years, the need for a unified clinicaltrial management platform has become increasingly evident.
In the cosmetics industry, innovation is key to staying ahead of the competition. As consumer preferences evolve and regulatory demands increase, cosmetics companies are under pressure to prove the safety and efficacy of their products through rigorous clinical studies.
This is where unified clinicaltrial management solutions come into the picture, enhancing efficiency, reducing time to market, and ensuring high-quality, scientifically validated consumer products. A delay in trials or approval can significantly impact the product’s success and the company’s bottom line.
Part 2 of this series will be devoted to clinical decision support (CDS) software. The FDA’s General Approach to Regulating mHealth Products. Food and Drug Administration (FDA) regulation as a medical device. If an mHealth device is life sustaining, then the FDA is certainly going to regulate the device.
Like with small molecules, clinicaltrials of biologics are designed to determine pharmacokinetics (PK), pharmacodynamics (PD), safety, and efficacy. The regulations regarding BLAs for therapeutic biological products are included in 21 CFR parts 600 , 601 , and 610. Expanded access to experimental biologics. BLA process (CBER).
Criteria for regulation. The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). peer-reviewed clinical studies or clinical practice guidelines) meet Criterion 2 and are not medical devices subject to FDA regulation and oversight.
Since that time, it was formalized in FDA regulations (21 CFR 314 Subpart H) in 1992, codified in the Food, Drug, & Cosmetic Act by FDAMA (21 USC 356(c)) in 1997, revised by FDASIA in 2012, and described in guidance, most importantly, in the 2014 Expedited Programs for Serious Conditions Drugs and Biologics (2014 Guidance).
On April 28, FDA issued its first Notice of Noncompliance for failure to submit required clinicaltrial results information to ClinicalTrials.gov. The “responsible party” is the sponsor or principal investigator of the trial. The notice was given to Acceleron Pharma, Inc., 42 C.F.R. §§ 11.10, 11.44(a). 42 U.S.C. §
As electronic documentation usage increases in clinicaltrials, organizations must make sure they’re still protecting participants’ health and safety without sacrificing quality or efficacy. One way to do so is to ensure the software platforms you are using to conduct research are in line with federal regulations.
As it becomes more complex with growing volumes of data, evolving regulations, and the pressure for faster drug development, traditional methods of clinical research management are no longer sufficient. Let’s delve into why this method is paramount for the transformation of clinical research management.
Conducting clinicaltrials to see if a drug is safe and effective can be a very time-consuming process. To meet the urgent demand for effective therapies, FDA has worked with clinicaltrial experts to rapidly advance best practices in the design and execution of clinicaltrials. Source link.
The letter , which was sent on January 4, 2021, stated that several of the company’s products violated section 505(a) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), 21 U.S.C. § 355(a) which prohibits the introduction and delivery of new drugs without the Secretary of Health of Human Service’s approval.
And if you don’t have the infrastructure and [a team adequately equipped] to run trials, it really is difficult to maintain doing trials.” Dr. Eder practices at Velocity’s research site in Binghamton that partners with United Health Services (UHS) to conduct clinicaltrials for a broad range of investigational products.
Butler — On April 28, FDA issued its first Notice of Noncompliance for failure to submit required clinicaltrial results information to ClinicalTrials.gov. The “responsible party” is the sponsor or principal investigator of the trial. The notice was given to Acceleron Pharma, Inc., 42 C.F.R. §§ 11.10, 11.44(a). 42 U.S.C. §
A PMR is a study “that sponsors are required to conduct under one or more statutes or regulations,” whereas a PMC is a study “that a sponsor has agreed to conduct, but that are not required by a statute or regulation” (see FDA Webpage, Postmarketing Requirements and Commitments: Introduction ).
Warning Letters, generally made public in a batch each Tuesday, are FDA’s public sanction that is most widely used to bring pressure on manufacturers and clinicaltrial investigators. Patcos Cosmetics Pvt. If FDA wanted to create that impression, it likely succeeded. They have great impact on most companies that receive them.
Given the similar course of COVID-19 disease in adults and pediatric patients, today’s approval of Veklury in certain pediatric patients is supported by efficacy results from phase 3 clinicaltrials in adults. Information on the trials in adults can be found in the FDA-approved drug labeling for Veklury. Source link: [link].
At the time we posted about the Jarkesy decision, we predicted that it would impact FDA-regulated industry: The impact on FDA matters could be significant. The Court reasoned that the SEC’s civil penalties “are designed to punish and deter, not compensate,” and that they are a remedy “that could only be enforced in courts of law.”
As part of its rolling submission, the company recently notified the agency of additional findings from its ongoing clinicaltrial. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Because anabolic steroids are also abused to enhance athletic performance and increase muscle strength, Congress has enacted three laws regulating anabolic steroids: the Anabolic Steroid Control Acts of 1990 and 2004, and the Designer Anabolic Steroid Control Act of 2014 (“DASCA”). at 50,040 codified at 21 C.F.R.
We use the term “confirmatory evidence” as short-hand for one of the two ways explicitly defined in the Federal Food Drug & Cosmetic Act (“FD&C Act”) for demonstrating substantial evidence of effectiveness. Here, FDA cited to survival data from the same clinicaltrial population as confirmatory evidence.
Brenzavvy’s approval was based on an impressive amount of data collected from 23 clinicaltrials involving over 5,000 patients with type 2 diabetes. The Federal Food, Drug, and Cosmetic Act (FFDCA) gives the FDA the legal authority to approve drugs for both humans and animals.
While there are currently six biosimilars for AbbVie’s Humira (adalimumab) that have been approved by the regulator, the company’s patents prevent biosimilars from being launched until 2023. The regulator allows biosimilars to show slight differences in clinically inactive components of a product.
A PANDA, or the Pre-Hatch-Waxman Abbreviated New Drug Application, refers to abbreviated drug applications submitted and approved under sections 505(b) and 505(c) of the Federal Food, Drug, and Cosmetic Act (FDCA) prior to the enactment of the Hatch-Waxman Amendments in 1984, as FDA announced in the Federal Register last week.
PAXLOVID is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of PAXLOVID under 564(b)(1) of the Food Drug and Cosmetic Act, unless the authorization is terminated or revoked sooner. There are limited clinical data available for PAXLOVID. Herbal Products: St.
effective in preventing COVID-19 disease among these clinicaltrial participants with 11 cases of COVID-19 in the vaccine group and 185 in the placebo group. The pharmacovigilance plan includes a plan to complete longer-term safety follow-up for participants enrolled in ongoing clinicaltrials. The vaccine was 94.1%
The sponsor is the pharmaceutical company conducting the trial. If you mean using a different contract research organization (CRO) for the different phases of clinicaltrials – that’s different. The standard for the audit used depends on the type of vendor and would include the applicable regulations and ICH guidelines.
In a clinicaltrial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. View original content to download multimedia: [link].
.
This device was studied in a 30-day randomized, sham-controlled trial of 70 patients. A sham therapy is an inactive treatment or procedure that is intended to mimic as closely as possible a therapy in a clinicaltrial. Patients in the sham group wore the device, but no vibratory stimulation was provided.
The EUA for bebtelovimab is supported by clinical and nonclinical data. The clinical data are from a phase 2, randomized, single-dose clinicaltrial evaluating the efficacy of bebtelovimab alone and bebtelovimab combined with other monoclonal antibodies for treating mild to moderate COVID-19. Source link: [link].
– Full-Year Global Net Revenues from the Aesthetics Portfolio Were $2.590 Billion; Global Botox Cosmetic Net Revenues Were $1.112 Billion. Global Botox Cosmetic net revenues were $493 million , an increase of 9.1 – Reports Fourth-Quarter Diluted EPS of $0.01 percent on a reported basis, or 45.0 Recorded a $4.7
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. As part of the Accelerated Approval, Oncopeptides was required to conduct “further adequate and well-controlled clinicaltrials to verify and describe clinical benefit.” That did not happen.
PAXLOVID is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of PAXLOVID under 564(b)(1) of the Food Drug and Cosmetic Act, unless the authorization is terminated or revoked sooner. There are limited clinical data available for PAXLOVID. Herbal Products: St.
In September 2022, Roche acquired Good Therapeutics for an upfront payment of $250 million, and has access to their PD-1-regulated IL-2 program. 2) Botox Therapeutic/Cosmetic Botox, or botulinum neurotoxin, is a neurotoxic protein produced by the Clostridium botulinum bacteria. in January 2023, according to the manufacturer.
PAXLOVID is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of PAXLOVID under 564(b)(1) of the Food Drug and Cosmetic Act unless the authorization is terminated or revoked sooner. There are limited clinical data available for PAXLOVID. Herbal Products: St.
Lenz, Principal Medical Device Regulation Expert — FDA’s Office of Women’s Health (OWH) recently celebrated its 30th anniversary. This office was formed in 1994 to promote the inclusion of women in clinicaltrials and to provide leadership on topics related to the health of women.
The safety and efficacy of the One Male Condom was studied in a clinicaltrial comprised of 252 men who have sex with men and 252 men who have sex with women. When used during anal intercourse, the One Male Condom should be used with a condom-compatible lubricant. All participants were between 18 through 54 years old.
Purdue pleaded guilty to one count of dual-object conspiracy to defraud the United States and to violate the Food, Drug, and Cosmetic Act, and two counts of conspiracy to violate the Federal Anti-Kickback Statute.
By Véronique Li, Senior Medical Device Regulation Expert — At the latest MedTech conference held in Boston, MA from October 24-26, we heard from FDA officials and industry representatives on a range of topics. Below, we provide a snapshot of the three-day event: Update on the International Medical Device Regulators Forum (IMDRF).
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content