This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The US Food and Drug Administration (FDA) has approved AbbVie’s Rinvoq (upadacitinib) for patients with Crohn’s disease who do not respond to TNF blockers, a common immune suppressant treatment for the condition. Data from two Phase III studies, U-EXCEED and U-EXCEL, involving 857 patients was used to support the approval.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinicaltrials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
The COVID-19 pandemic has catalysed significant changes in the way pharma develops drugs, particularly in the clinicaltrial space. Hybrid or decentralised clinicaltrials (DCTs) have gained traction as technology, infrastructure and knowledge have evolved to support their use. Source: Izmailova et al, 2017.
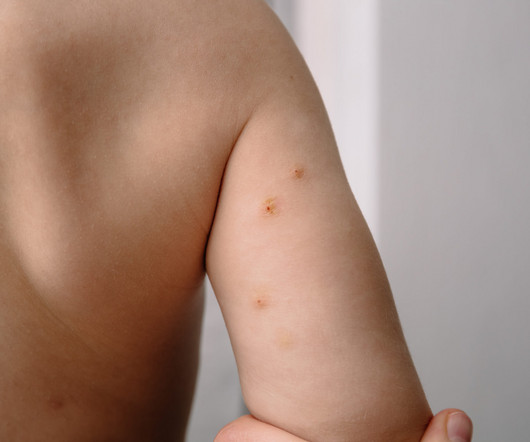
has received US Food and Drug Administration (FDA) approval for treating molluscum contagiosum in adult and pediatric patients aged two years and older in the US. Formerly known as VP-102, Ycanth is the first cantharidin formulation approved for this purpose. Another noteworthy contender is Novan Inc.’s
Daxxify was FDA-approved for similar cosmetic purposes as Botox and other neuromodulators like Dysport and Xeomin. Used for both cosmetic and therapeutic cases, Botox is a US Food and Drug Administration (FDA)-approved injection of botulinum toxin, a neurotoxic protein that can effectively paralyze the facial muscles.
For instance, Vyjuvek , the first FDA-approved gene therapy for DEB, is priced at $24,250 per vial. The clinical study also highlighted the favorable tolerability profile of Filsuvez. The most commonly reported adverse reactions in the clinicaltrial were pruritus (itching) and pain at the wound application site, occurring in 7.3
XTALKS WEBINAR: All Means All: The Road to Inclusivity in ClinicalTrials. Register for this free webinar to learn what the FDA’s new draft guidance means for diverse and inclusive trials. ClinicalTrials of Vtama. Live and On-Demand: Tuesday, July 12, 2022, at 11am EDT (4pm BST/UK).
Eichenfield serves on Verrica Pharmaceuticals’ Board of Directors and spoke to Xtalks about the company’s recent approval of YCANTH (cantharidin) topical solution as the first FDAapproved treatment for pediatric and adult patients with molluscum contagiosum, a highly contagious viral skin infection that primarily affects children.
Incytes recent announcement on the topline results from two Phase III clinicaltrials of povorcitinib in patients with hidradenitis suppurativa has stirred both hope and caution in the market. Janus kinase one (JAK1) plays a central role in the signaling pathways of several inflammatory cytokines.
Presented at the European Academy of Dermatology and Venereology (EADV) Congress 2024, the study highlighted significant improvements in key disease markers, positioning orismilast as a potential breakthrough in atopic dermatitis management.
In clinicaltrials, patients experienced significantly clearer skin and less interference with sleep due to itch when taking lebrikizumab compared to placebo. The ADvocate1 and ADvocate2 Phase III studies were published in the New England Journal of Medicine and the British Journal of Dermatology , respectively.
The approval of Sofdra provides a much needed solution for individuals struggling with this socially and physically debilitating condition. Hyperhidrosis is the third most prevalent dermatological condition in the US, affecting roughly 15.3 million people, following acne and atopic dermatitis.
FDAapproves Pfizer’s LITFULO™ (ritlecitinib) for adults and adolescents with severe alopecia areata Pfizer Inc. Food and Drug Administration (FDA) has approved LITFULO™ (ritlecitinib), a once-daily oral treatment, for individuals 12 years of age and older with severe alopecia areata. Source link: [link]
30, 2020 /PRNewswire/ — Eli Lilly and Company (NYSE: LLY) and Incyte (NASDAQ: INCY) announced today new data for baricitinib (marketed as OLUMIANT ® ) will be presented at the annual Fall ClinicalDermatology meeting taking place virtually October 29-November 1, 2020. INDIANAPOLIS , Oct.
FDAapproval based on positive results of international, multi-center ProDERM study. The prospective, double-blind, placebo-controlled Phase III clinicaltrial enrolled 95 patients at 36 sites globally, including 17 sites in the U.S., Credit: Octapharma. PARAMUS, N.J. – The U.S. 1, 2, 3, 4. Original Source.
Leqselvi’s approval is based on data from two Phase III clinicaltrials, THRIVE-AA1 and THRIVE-AA2, which together enrolled over 1,200 patients. Leqselvi works by selectively inhibiting the Janus kinases (JAK) JAK1 and JAK2, pathways that play a key role in this misguided immune response.
Sanofi said it would also apply its mRNA vaccine platform – acquired along with Translate Bio earlier this year – to find other acne vaccine candidates that could start clinicaltrials in 2023. Origimm’s expertise in the skin microbiome and antigen discovery will be central to that effort, it added.
On September 1, 2022, Boehringer Ingelheim Pharmaceuticals announced in a press release that the US Food and Drug Administration (FDA) approved Spevigo (spesolimab-sbzo) intravenous injections for GPP flares in adults. Spevigo is the first approved treatment option for GPP in adults. ClinicalTrial Behind Spevigo.
percent ruxolitinib, a topical Janus kinase (JAK) inhibitor, is US Food and Drug Administration (FDA)-approved for the treatment of mild to moderate atopic dermatitis in patients 12 years and older. The initial FDAapproval for atopic dermatitis paved the way for the 2022 FDAapproval for vitiligo.
Last summer, Verrica Pharmaceuticals’ topical Ycanth (cantharidin) received FDAapproval as the first treatment for molluscum contagiosum. The FDA’sapproval of Zelsuvmi was based on data from two Phase III clinicaltrials, B-SIMPLE 4 and B-SIMPLE 2, involving 1,598 patients.
Fast Track designation is well-timed, as we anticipate starting our Phase 2 clinicaltrial in hospitalized COVID-19 patients this month, and should help bring Brilacidin to patients faster in these dire times.”. Brilacidin, a versatile compound with broad therapeutic potential, is in a new chemical class called defensin-mimetics.
In a major development for those battling moderate-to-severe atopic dermatitis, the US Food and Drug Administration (FDA) approved Eli Lilly’s Ebglyss (lebrikizumab-lbkz). The FDAapproval is based on data gathered in three pivotal Phase III clinicaltrials: ADvocate 1, ADvocate 2 and ADhere.
Emrosi’s approval was based on data from a pair of Phase III clinicaltrials for the treatment of rosacea. The trials met all co-primary and secondary endpoints, with participants having successfully completed the 16-week treatment with no significant safety issues.
The new findings from the Phase 3 clinicaltrials (ADvocate 1 and 2) showed eight out of ten patients who achieved clinical response (EASI-75*) with lebrikizumab monotherapy at 16 weeks maintained skin clearance at one year of treatment with the once every two weeks or four weeks regimen. . Almirall S.A.’s
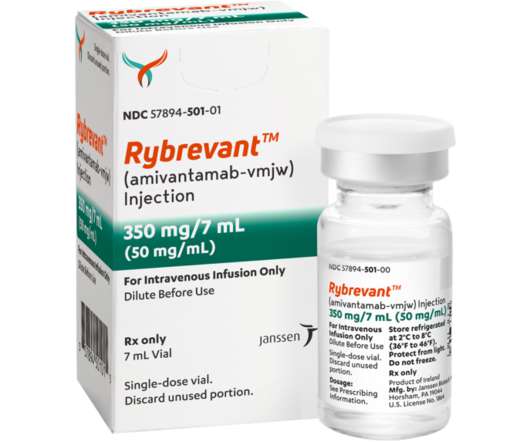
Food and Drug Administration (FDA) approval for patients with locally advanced or metastatic NSCLC with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, based on data showing an ORR of 40 percent (95 percent CI, 29 – 51) and median duration of response of 11.1 Dermatologic Adverse Reactions 7. of patients.
OLUMIANT is a once-daily, oral JAK inhibitor approved in the U.S. FDA-approved labeling for Olumiant includes a Boxed Warning for Serious Infections, Malignancy, and Thrombosis. Lymphopenia – Absolute lymphocyte count (ALC) <500 cells/mm 3 were reported in Olumiant clinicaltrials. About Lilly in Dermatology.
3 “Nivolumab provides a new FDA-approved treatment shown to reduce the risk of disease recurrence or death based on the safety and efficacy findings from CheckMate -274, and has the potential to become a new standard of care option in this setting.” Galsky,* M.D., 1 Please see Important Safety Information below.
This classification aims to expedite the development and review of drugs that are intended to treat a serious condition when preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over already available therapies on a clinically significant endpoint(s). Increases of ALT ?5x
Amgen’s Otezla (apremilast) continues to expand its reach, with the US Food and Drug Administration (FDA) approving its use for children and adolescents with moderate to severe plaque psoriasis in 2024. This drug may significantly improve psoriasis care by offering effective long-term control with fewer side effects.
Food and Drug Administration (FDA) approved SKYRIZI 150 mg in April based on data from three clinicaltrials showing the single-dose SKYRIZI 150 mg injection was bioequivalent, working the same as two injections of SKYRIZI 75 mg per dose with a consistent efficacy and safety profile. ” The U.S.
Opdivo ’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology, and includes a broad range of clinicaltrials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients.
Opdivo ’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology, and includes a broad range of clinicaltrials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients.
Opdivo ’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology, and includes a broad range of clinicaltrials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients.
Paxlovid was first authorized under the FDA Emergency Use Authorization in December 2021 ; however, it has received FDAapproval on May 25, 2023. pneumoniae serotypes) replaced the company’s first pneumococcal conjugate vaccine Prevnar (PCV7, approved by the FDA in February 2000) in a February 2010 FDAapproval.
Opdivo ’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology, and includes a broad range of clinicaltrials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients.
Allergan Aesthetics, an AbbVie company renowned for its development of leading aesthetics brands and products, recently announced the US Food and Drug Administration (FDA) approved their ground-breaking hyaluronic acid product, SkinVive by Juvéderm, for achieving remarkable skin smoothness.
RELATED: Innovations and ClinicalTrial Diversity in Medical Aesthetics: Insights from Dr. Stephanie Manson Brown, VP & Head of Clinical Development at Allergan Aesthetics – Xtalks Life Science Podcast Ep. 134 How Does Letybo Work?
The US Food and Drug Administration (FDA) has approved Galderma’s monoclonal antibody Nemluvio (nemolizumab) for the treatment of adult patients with the chronic skin condition prurigo nodularis. A significant percentage of patients also reported reduced sleep disturbances by Week 16 in both clinicaltrials.
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. Food and Drug Administration (FDA) has approved Klisyri (tirbanibulin) for the topical treatment of actinic keratosis (AK) on the face or scalp. The FDAapproval of Klisyri is a significant milestone for Athenex.
The US Food and Drug Administration (FDA) has approved Iwilfin (eflornithine) to reduce the risk of relapse in adult and pediatric patients with high-risk neuroblastoma (HRNB) who have had at least a partial response to previous multiagent, multimodality therapy including anti-GD2 immunotherapy.
There are currently no FDA-approved treatment options for EB, a rare group of inherited disorders characterised by extremely fragile skin that blisters and tears from even minor friction or trauma. pic.twitter.com/NzhGx1q1o8. — debra of America (@debraOfAmerica) February 28, 2022.
And this year Eli Lilly stepped into the obesity drug market with its own GLP-1 weight loss drug Zepbound (tirzepatide), which the US Food and Drug Administration (FDA) approved in November of this year. There has been significant growth in the number of registered clinicaltrials globally, with a focus on interventional studies.
Immune-mediated adverse reactions can occur at any time during or after treatment with KEYTRUDA, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, dermatologic reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplantation. Nephritis resolved in 56% of the 9 patients.
An IQVIA survey to Neurology, Cardiology, Rheumatology, Dermatology and Ophthalmologists in June 2020 across the lead five European countries showed an average of 30% of patients either “no shows” or still waiting for treatment that was delayed.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






































Let's personalize your content