This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The American Conference Institute (“ACI”) is holding its 2nd West Coast Editionof its Legal, Regulatory, and Compliance Forum on Cosmetics & Personal Care Products from September 25-26 at the Le Meridien Delfina, Santa Monica, California. s John W.M. s John W.M. s John W.M.
As readers of our blog know, MoCRA was a significant change to regulation of cosmetics. Now in the second year of implementation, companies have started noticing the consequences as FDA implements the new requirements and develops regulations and guidance. By John W.M. Truly, these are complex considerations.
In the cosmetics industry, innovation is key to staying ahead of the competition. As consumer preferences evolve and regulatory demands increase, cosmetics companies are under pressure to prove the safety and efficacy of their products through rigorous clinical studies.
Importantly, the 2018 Farm Bill preserved FDA authority to regulate products with cannabis or cannabis-derived compounds under the Federal Food, Drug, and Cosmetic (FD&C) Act and Section 351 of the Public Health Service Act. The post CBD Research: A Dive into the Regulations of Cannabis Research appeared first on Advarra.
The question that is always on the mind of folks in FDA-regulated industries is, what does that mean for my application/inspection/meeting? The answers below are based on my experience as a reviewer and compliance officer at FDA during the 5-week 2018/2019 government shutdown. What does it mean for FDA staff?
devices must be regulated as devices, and drugs—if they do not also satisfy the device definition—must be regulated as drugs.”. PANDAs have historically been overseen by the FDA’s Office of Generic Drugs and thus regulated more like a generic than an NDA. Final Regulation Issued for “Intended Use”.
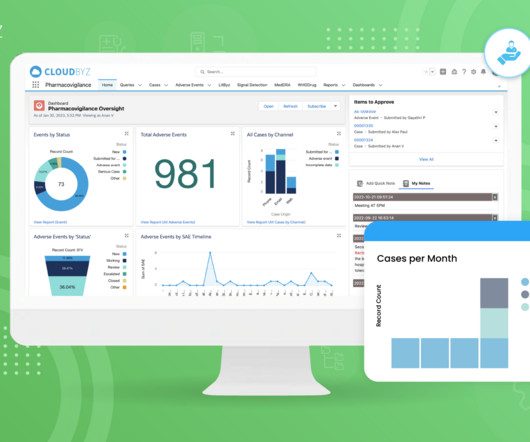
The cosmetics and personal care industry is one where customer trust is paramount, given that the products are applied directly to the skin, hair, and body. A well-implemented safety vigilance solution ensures that companies stay compliant with these regulations, minimizing the risk of sanctions or penalties.
One way to do so is to ensure the software platforms you are using to conduct research are in line with federal regulations. In 1997, FDA released regulations providing guidance on the use of electronic systems. Un-specified identified Agency regulations. There are many factors in how Part 11 regulations affect software.
Walsh — Among FDA-regulated establishments and stakeholders, there is one word that makes everyone go on edge – the dreaded FDA “inspection.” Document all corrective actions and follow-up to ensure sustained compliance. By Sarah Wicks & Anne K. In FY2023, FDA conducted over 1000 inspections under the BIMO program.
The FDA’s General Approach to Regulating mHealth Products. Food and Drug Administration (FDA) regulation as a medical device. Determining whether a product is a medical device subject to FDA regulation necessarily begins with understanding the FDA regulatory definition of ‘medical device’. not a medical device, ii.
Mullen — On February 23, 2022, FDA published in the Federal Register a proposed rule that would replace the Quality System Regulation (QSR), at 21 C.F.R. Part 820, with a newly named Quality Management System Regulation (QMSR). ISO 13485, while largely duplicative to the QSR, is not a perfect fit with other existing FDA regulations.
Circuit , FDA published a Federal Register Notice today (August 9) soliciting comments on its proposed approach to implementing the Court’s interpretation of the Federal Food, Drug, and Cosmetic Act (FDCA) distinction between drugs and devices.
FDA Inspections - Overview admin Thu, 02/02/2023 - 13:52 FDA Inspection is a regulatory process conducted by the United States Food and Drug Administration (FDA) to evaluate the compliance of food and drug establishments with FDA regulations and standards.
The discussion paper provides an overview of FDA’s current approach to regulation of 3D-printed devices. In brief, such devices can be commercially distributed to the general public for non-medical purposes without FDA regulation (e.g., use in education, construction, art, and jewelry). products intended for medical purposes).
By Riëtte van Laack — FDA regulates pet food similar to other animal foods. The Federal Food, Drug, and Cosmetic Act (FDC Act) requires that all animal foods, like human foods, be safe to eat, produced under sanitary conditions, contain no harmful substances, and be truthfully labeled.
Baumhardt , MS, MJ, MT(ASCP), RAC, FRAPS, has joined the firm as a Senior Medical Device Regulation Expert, and that Sophia Gaulkin has joined the firm as an Associate. Baumhardt advises clients on complaint handling, MDRs, Quality System Regulationcompliance and enforcement matters. Hyman, Phelps & McNamara, P.C. (“HP&M”)
The American Conference Institute (“ACI”) will be hosting a series of go-to forums on critical topics including novel therapeutics, cosmetics/personal care products and Paragraph IV disputes. Claud will be featured at the Legal, Regulatory, and Compliance Forum on Cosmetics & Personal Care Products in New York, NY.
Criteria for regulation. The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). peer-reviewed clinical studies or clinical practice guidelines) meet Criterion 2 and are not medical devices subject to FDA regulation and oversight.
To help ensure compliance, it is advisable for companies to establish procedures to assess and verify IT security of social media and digital channels, and to review and monitor company activities, content, and materials. Why is guidance needed? What are the rules on Social Media Influencers and Digital Opinion Leaders? About the authors.
Claud is a 15-year veteran of the Department of Justice, where most recently he was an Assistant Director of the Consumer Protection Branch overseeing the Corporate Compliance and Policy Unit. He is a frequent public speaker on matters of government enforcement strategies under the FDCA as well as corporate compliance best practices.
With the approach of the January 1, 2022 mandatory compliance deadline for the BE standard, manufacturers and importers of food and dietary supplements should work to develop strategies for compliance and evaluating each product’s bioengineered (BE) status if they have not already done so.
The American Conference Institute (“ACI”) will be hosting the go-to forum for critical updates on OTC regulation and enforcement, monograph reform, ACNU and advertising essentials… and FDA Law Blog readers can get a discount. Deb along with fellow panelists Kyle Y.
The discussion paper provides an overview of FDA’s current approach to regulation of 3D-printed devices. In brief, such devices can be commercially distributed to the general public for non-medical purposes without FDA regulation (e.g., use in education, construction, art, and jewelry). products intended for medical purposes).
The 21 st Century Cures Act generated a good deal of excitement and interest when it added a section called “ Utilizing Real World Evidence ” to the Food, Drug, and Cosmetic Act (Section 505F). Manager, Regulatory Compliance. FDA Guidance Addresses Real-World Evidence Data Standards. Co-Authors: Bill Stoltman, JD. Jenny Fielder.
Whether Florida is able to import drugs from Canada, including in light of Canadian regulations controlling drug exports, remains to be seen. As we reported , the export regulations first issued as an interim order in November 2020, in response to the FDA publishing the US Importation Rule in October 2020. 01.014.13).
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
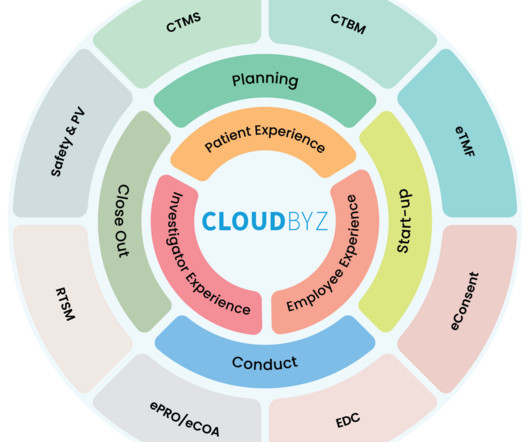
Regulatory Compliance: Streamlining processes aids in meeting stringent regulatory requirements and maintaining data integrity. Conclusion A unified clinical trial management platform is no longer a luxury but a necessity for pharmaceutical companies aiming to thrive in an increasingly competitive and regulated industry.
Mullen — On January 18, 2024, the director of FDA’s Center for Devices and Radiological Health and the chief medical officer and acting director of CMS’ Center for Clinical Standards and Quality issued a joint press release supporting FDA’s recent proposed rule regulating Laboratory Developed Tests (LDTs).
an animal food manufacturing company of Carney’s Point, New Jersey, has agreed to stop selling, manufacturing and distributing raw pet food and come into compliance with the Federal Food, Drug, and Cosmetic Act (FD&C Act). Food and Drug Administration (FDA) announced today that Bravo Packing, Inc., Related Information.
By Véronique Li, Senior Medical Device Regulation Expert & Ana Loloei & Allyson B. Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. Revised § 820.3
filed comments on behalf of the Coalition to Preserve LDT Access and Innovation in response to FDA’s proposed rule to regulate laboratory developed tests (LDTs) as devices. Weighing in at nearly 60 pages, the comments detail extensive flaws in the proposed regulation. Javitt — On Monday, Hyman, Phelps & McNamara, P.C.
Those who attended their annual Enforcement, Litigation, and Compliance Conference holiday reception earlier this month got a sneak peek at this new branding effort with the unveiling of the association’s new logo ( here ). 2024 brings a host of Hyman, Phelps & McNamara, P.C. (“HPM”)
Field preemption applies when federal regulation occupies the regulated area so pervasively that it leaves no room for states to supplement it, even if the state law is consistent with the federal law. The court also noted that the practice of pharmacy is an area traditionally left to state regulation.
As it becomes more complex with growing volumes of data, evolving regulations, and the pressure for faster drug development, traditional methods of clinical research management are no longer sufficient. Integrated Compliance and Governance With stringent regulations governing clinical trials, compliance is of utmost importance.
deputy director of the FDA’s Center for Veterinary Medicine (CVM) Division of Compliance: “Although this pet food recall is still unfolding, we are sharing the facts we have so far because the levels of aflatoxin found in the recalled pet food are potentially fatal. SILVER SPRING, Md. , SOURCE U.S. Food and Drug Administration.
The trials span several categories of consumer products such as cosmetics, personal care products, nutrition supplements, food, beverages, and consumer health products. This includes trial design, participant recruitment, data collection and analysis, and regulatory compliance.
Food and Drug Administration (FDA) released a draft update to its Compliance Policy Guide (CPG) for FDA staff on the Agency’s enforcement of major food allergen labeling and cross-contact. Gaulkin & Riëtte van Laack — On May 16, the U.S.
that are not in compliance with applicable requirements. These proposed regulations do not include a prohibition on individual consumer possession or use. Source link: [link].
The amended regulation would read as follows (revisions underlined): In vitro diagnostic products are those reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae.
Shapiro — After 23 years, de novo classification review finally has an implementing regulation ! The other major review processes have had their regulations in place for many decades. Only a year later, in 1977, FDA promulgated regulations governing 510(k) reviews (21 C.F.R. Part 807, Subpart E).
However, many details remain to be addressed in forthcoming regulations. CIPO consulted on proposed features of the regulations in 2023 (see here ), including many factors that would reduce the length of the PTA term. Draft regulations are expected to be released for consultation in 2024. The Guidance was last updated in 2005.
The LMG awards are reflective of HPM’s excellence across several of our core life sciences practices, from controlled substances work to drug development, to Hatch-Waxman, to drug pricing, to medical device regulation. That combined depth and breadth of experience is why clients come to us.
During the examination, the FDA investigator made several compliances that are outlined in an examination bonus report, also known as an “ FDA Form 483.” The FDA investigator handed a list of their compliances to the company. Source link: [link].
As we outlined in the Closer to Zero action plan, the agency is increasing targeted compliance activities as part of our efforts to monitor levels of these elements in foods through the FDA’s Total Diet Study , Toxic Elements in Food and Foodware program and sampling assignments,” said Susan Mayne, Ph.D., Source link: [link].
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content