This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Although mHealth has been gaining in popularity for at least the past decade, before commercializing their mHealth products, developers must determine whether the product is subject to U.S. If so, developers must develop and execute on a regulatory strategy. The FDA’s General Approach to Regulating mHealth Products.
Training and development. Staff development and training programmes were largely on pause during the pandemic. Businesses need to ensure they prioritise the recruitment and development of staff to help ensure longevity and survival. Harness technology.
Such non-device CDS are: (1) not intended to acquire, process, or analyse a medical image or a signal from an in vitro diagnostic device or a pattern or signal from a signal acquisition system (Criterion 1); (2) intended for the purpose of displaying, analysing, or printing medical information about a patient or other medical information (e.g.
Mullen — Happy Birthday Laboratory Developed Tests (LDTs). If anything, the announcement was remarkably inconspicuous: a seemingly throwaway sentence in a draft compliance policy guide for research use only and investigational use only products. As with any “child” they’ve matured and developed as they’ve grown into adulthood.
The first phaseout milestone is less than a year away; by May 6, 2025 most laboratories will need to demonstrate compliance with Medical Device Reporting (21 C.F.R. § 803), Reporting of Corrections and Removals (21 C.F.R. § 806) and Complaint Files (21 C.F.R.
Manufacturers must adhere to a wide array of standards and guidelines that dictate how devices are developed, tested, and brought to market. Compliance with their regulations is mandatory for manufacturers seeking to market their devices globally. Regulatory bodies such as the U.S. What is the Medical Device Safety process?
Vaccines are our number one weapon in the fight against infectious diseases, but their development has historically involved a long and complex process taking up to a decade. Before COVID-19, Merck held the record for the fastest modern vaccine ever developed. Single- and multi-nation funding opportunities flooded in.
Feature articles during April focused on the regulatory profession and specifically, professional development and career advancement. Continuous professional development. The results demonstrate that regulatory knowledge and qualifications are an advantage and of value in personal and professional development.
The main purpose of in vitro diagnostic solutions is to perform non-invasive / minimally invasive test procedures in order to detect a variety of health conditions using samples, such as blood, tissue or urine. Such tests help to diagnose health conditions, monitor treatments, perform disease screening and aid in early diagnosis of diseases.
Loloei tackled legal matters related to various aspects of the regulation of medical devices, in vitro diagnostics, and combination products including regulatory and compliance issues, dispute resolutions between the FDA and sponsors, and FDA enforcement actions. While at FDA, Ms. During her FDA tenure, Ms. Gibbs , HP&M Director.
Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ).
BetterLife” or the “Company”) (CSE: BETR / OTCQB: BETRF / FRA: NPAU ), an emerging biotech company focused on the development and commercialization of cutting-edge treatments in mental disorders and viral infections, announces it has entered into an agreement with Eurofins Discovery for TD-0148A’s U.S. VANCOUVER, Jan.
To help ensure compliance, it is advisable for companies to establish procedures to assess and verify IT security of social media and digital channels, and to review and monitor company activities, content, and materials. What is the scope of the Guidance? Companies can sponsor website content if the role of the company is made clear.
Baumhardt provides counsel to medical device, in vitro diagnostic, and combination product manufacturers on a wide range of pre- and post-marketing regulatory topics. Baumhardt advises clients on complaint handling, MDRs, Quality System Regulation compliance and enforcement matters. In the pre-market area, Ms. In addition, Ms.
Javitt & Philip Won — As we reported last week, FDA has issued a 26 page, single spaced, tiny-font Proposed Rule of Laboratory Developed Tests (LDTs). There is much to unpack, and we intend to do so in a series of blog posts. In this post, we focus on the proposed changes themselves, and the many questions the agency leaves unanswered.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). We take this date with a grain of salt given the frequency with which these dates are missed and given the volume of comments filed. (It
But to meet aggressive milestones and stringent performance standards, there’s a need for continuous innovation and streamlined development and quality control workflows. Sartorius is a globally recognized player in the diagnostics industry, providing a variety of solutions for in vitro diagnostics kit (IVD) manufacturers.
Streamlined in vitro data, published in bioRxiv, demonstrate that sotrovimab retains exertion against all current variants of concern and interest of the SARS-CoV-2 contagion as defined by the World Health Organization, plus others, including, but not limited to, Delta (B.1.617.2), 1.617.2), Delta Plus (AY.1 2) and Mu (B.1.621).
Complaints FDA discussed how LDT developers should handle the transition from the current Quality System Regulation (QSR)(21 CFR Part 820) to the recently promulgated Quality Management System Regulation (QMSR) that is scheduled to take effect on February 2, 2026. SCF : LDT, unmet need within an integrated healthcare system.
The test is planned for commercial launch as a CE-IVD ( in vitro diagnostic) certified product in the European Union in Q1 2021. “Applying 3a’s proprietary enhanced RNA technology to PCR testing was a logical next step in our product development pipeline,” said Dr. .
It has become the de facto standard equipment for high titer transient protein expression platforms used by many leading Chinese antibody drug pharmaceutical companies and IVD (In Vitro Diagnostics) companies since its launch in Oct. Etta Biotech is a leading cell electroporation technology and equipment supplier. JS Bio’s parent company.
In vitro diagnostic (IVD) devices are tests used on human biospecimens (e.g., IVDs meeting this definition are known as laboratory developed tests (LDTs). The developer/manufacturer of the IVDs are typically the entity that submits the study. IVDs are used in almost all clinical research.
filed comments on behalf of the Coalition to Preserve LDT Access and Innovation in response to FDA’s proposed rule to regulate laboratory developed tests (LDTs) as devices. As a threshold matter, FDA lacks the power to regulate tests developed and used in a laboratory. Javitt — On Monday, Hyman, Phelps & McNamara, P.C.
Regulations for research involving devices, in vitro diagnostics (IVDs), and digital therapeutics differ from those governing pharmaceutical development. In other words, what are the risks and benefits of the product? The greater the risk, the more likely a series of trials for market clearance or approval will be required.
If a new medical device, software as a medical device (SaMD), or in vitro diagnostic (IVD) is poised to successfully make it through the development process to commercialization, sponsors must consider a trifecta of interconnected systems involving: Regulatory strategy and submissions. Quality assurance and risk management.
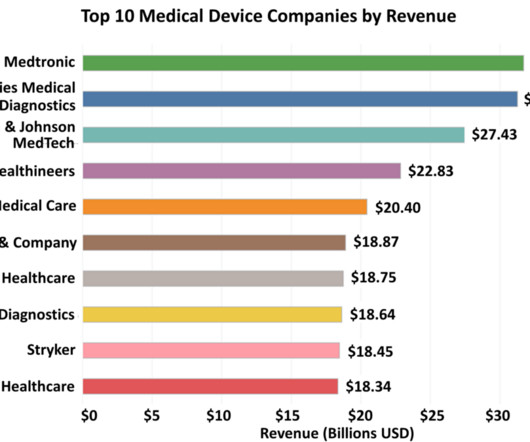
These powerhouse organizations have not only distinguished themselves through their cutting-edge devices and technologies but also via their robust research and development (R&D) capabilities, unwavering commitment to quality and extensive global reach. 1) Medtronic 2022 Revenue: Medtronic reported an annual revenue of $31.69
Mullen — On January 18, 2024, the director of FDA’s Center for Devices and Radiological Health and the chief medical officer and acting director of CMS’ Center for Clinical Standards and Quality issued a joint press release supporting FDA’s recent proposed rule regulating Laboratory Developed Tests (LDTs).
Clinical trials involving investigational in vitro diagnostic (IVD) devices are subject to the Food and Drug Administration’s (FDA’s) investigational device exemption (IDE) requirements ( 21 CFR 812 ). When an investigational device does not carry this potential for serious risk, the clinical investigation is NSR.
The final guidance therefore may have significant implications for a wide range of stakeholders, including not only software developers but also health care providers, hospitals, patients and payors. Criterion 2 : Non-Device CDS software functions display, analyze or print medical information about a patient or other medical information.
CENTOGENE’s SARS-CoV-2 test is a molecular diagnostic test performed for the in vitro qualitative detection of RNA from the SARS-CoV-2 in oropharyngeal samples from presymptomatic patients and probands according to the recommended testing by public health authority guidelines.
CAMBRIDGE, Mass. and ROSTOCK, Germany and BERLIN, Nov.
Bacteria plays a crucial role in maintaining the ecosystem balance. However, there are few species of bacteria that can cause several infectious diseases ( such as strep throat, salmonellosis, tuberculosis, whooping cough ). These are mainly transmitted through air, water, living organisms, and food.
Overall, patient compliance for PROs was greater than 90 percent. The PRO analysis included patients in both arms of the study and measured their experiences with side effects, symptoms, and health-related quality of life, in those receiving Verzenio plus ET versus ET alone. The detailed data were presented at the virtual 17th St.
Gibbs The multi-decade battle over FDAs power to regulate Laboratory Developed Tests (LDTs) had its day in court earlier this week. Citing the May 6 compliance date for Stage 1 of the LDT Rule, both ACLA and AMP asked that Judge Jordan issue his ruling expeditiously. By Allyson B. Mullen & Jeffrey N.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.




































Let's personalize your content