This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The FDA’s General Approach to Regulating mHealth Products. Food and Drug Administration (FDA) regulation as a medical device. Determining whether a product is a medical device subject to FDA regulation necessarily begins with understanding the FDA regulatory definition of ‘medical device’. not a medical device, ii.
Baumhardt , MS, MJ, MT(ASCP), RAC, FRAPS, has joined the firm as a Senior Medical Device Regulation Expert, and that Sophia Gaulkin has joined the firm as an Associate. Baumhardt provides counsel to medical device, in vitro diagnostic, and combination product manufacturers on a wide range of pre- and post-marketing regulatory topics.
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
Criteria for regulation. The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). peer-reviewed clinical studies or clinical practice guidelines) meet Criterion 2 and are not medical devices subject to FDA regulation and oversight.
To help ensure compliance, it is advisable for companies to establish procedures to assess and verify IT security of social media and digital channels, and to review and monitor company activities, content, and materials. Why is guidance needed? What is the scope of the Guidance?
Loloei tackled legal matters related to various aspects of the regulation of medical devices, in vitro diagnostics, and combination products including regulatory and compliance issues, dispute resolutions between the FDA and sponsors, and FDA enforcement actions. While at FDA, Ms. During her FDA tenure, Ms.
The crux of the proposed rule lies in the addition of ten words: “ including when the manufacturer of these products is a laboratory.” These words would be added to the definition of “ in vitro diagnostic [IVD] products” in 21 C.F.R. There is much to unpack, and we intend to do so in a series of blog posts.
A proposal has been put forward to amend the transitional provisions for certain medical devices and in vitro diagnostic medical devices (amending Regulations (EU) 2017/745 (MDR) 1 and (EU) 2017/746 (IVDR) 2. – The extension is directly applicable, therefore changing the date on the individual certificates is not necessary.
1] Yet FDA’s conclusions about the Agency’s ability to regulate the entire laboratory industry are based on fundamentally flawed assumptions about the number of entities and tests that will be subject to FDA regulation. 5] This stratification, though, assumes that LDTs will follow the same pattern as IVDs currently regulated by FDA.
Complaints FDA discussed how LDT developers should handle the transition from the current Quality System Regulation (QSR)(21 CFR Part 820) to the recently promulgated Quality Management System Regulation (QMSR) that is scheduled to take effect on February 2, 2026. By Steven J. Gonzalez & Lisa M.
Eurofins Discovery will be conducting the IND-enabling in-vitro preclinical primary pharmacology and safety pharmacology studies on TD-0148A at its state-of-the-art facilities at Eurofins Cerep, DiscoverX and Panlabs. FDA Investigational New Drug (“IND”)-enabling pharmacology studies. About BetterLife Pharma: BetterLife Pharma Inc.
Mullen — On January 18, 2024, the director of FDA’s Center for Devices and Radiological Health and the chief medical officer and acting director of CMS’ Center for Clinical Standards and Quality issued a joint press release supporting FDA’s recent proposed rule regulating Laboratory Developed Tests (LDTs).
In vitro diagnostic (IVD) devices are tests used on human biospecimens (e.g., Additionally, while the Food and Drug Administration (FDA) regulates all IVDs as medical devices, the agency has generally not enforced device regulations for certain IVDs designed, manufactured, and used within a single CLIA-certified laboratory.
filed comments on behalf of the Coalition to Preserve LDT Access and Innovation in response to FDA’s proposed rule to regulate laboratory developed tests (LDTs) as devices. Weighing in at nearly 60 pages, the comments detail extensive flaws in the proposed regulation. Javitt — On Monday, Hyman, Phelps & McNamara, P.C.
If a new medical device, software as a medical device (SaMD), or in vitro diagnostic (IVD) is poised to successfully make it through the development process to commercialization, sponsors must consider a trifecta of interconnected systems involving: Regulatory strategy and submissions. Meeting with the Regulators. Intended use.
Regulations for research involving devices, in vitro diagnostics (IVDs), and digital therapeutics differ from those governing pharmaceutical development. Interpreting evolving regulations for these devices is often a unique challenge for emerging biotech companies. In other words, what are the risks and benefits of the product?
In other words, the FDA also regulates whether investigational products may be manufactured, shipped, and administered to human subjects who participate in clinical investigations. . The Food and Drug Administration (FDA) is the federal entity in the U.S. When Does an IND go into Effect? . The IND goes into effect; and . 21 CFR 312.40(a)
The test is planned for commercial launch as a CE-IVD ( in vitro diagnostic) certified product in the European Union in Q1 2021. 3a will continue to advance its industrial design, product usability, labelling compliance, regulatory approval, and production planning work in parallel to its validation studies. .”
Other articles examined communication – one on persuasive skills, another on disseminating of regulatory intelligence – and the regulatory response to nitrosamine contamination of drug products. . . Continuous professional development. DiMichele provides valuable information on some of the available programs. . .
The final CDS Guidance represents a marked change in approach from prior drafts and foreseeably will result in the regulation of many types of CDS that were previously considered to be Non-Device CDS or low-risk Device CDS under enforcement discretion. Medical information about a patient.
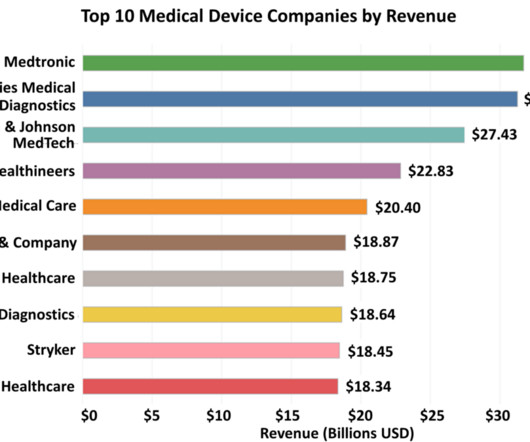
As technology continues to revolutionize every sector of our lives, the medical device industry stands at the forefront of this innovation, playing a pivotal role in enhancing patient care, improving diagnostic accuracy and transforming treatment modalities. Note: When it comes to companies that report in foreign currencies, the conversion to U.S.
CENTOGENE’s SARS-CoV-2 test is a molecular diagnostic test performed for the in vitro qualitative detection of RNA from the SARS-CoV-2 in oropharyngeal samples from presymptomatic patients and probands according to the recommended testing by public health authority guidelines.
CAMBRIDGE, Mass. and ROSTOCK, Germany and BERLIN, Nov.
Introduction to Medical Device Safety, Systems, and Regulations The medical device industry is a rapidly evolving field that plays a critical role in modern healthcare. This is where the importance of stringent safety processes, robust systems, and comprehensive regulations comes into play. What is the Medical Device Safety process?
Thirty years ago today, FDA announced that it had the authority to regulate you. If anything, the announcement was remarkably inconspicuous: a seemingly throwaway sentence in a draft compliance policy guide for research use only and investigational use only products. By Jeffrey N. Gibbs & Allyson B. 92P-0405 (Aug.
Laboratories struggling to understand the myriad implications of being regulated as device “manufacturers” were hopeful that additional guidance would shed light on how to apply FDA’s existing medical device regulatory framework to their operations. 803), Reporting of Corrections and Removals (21 C.F.R. § 806) and Complaint Files (21 C.F.R.
Sartorius is a globally recognized player in the diagnostics industry, providing a variety of solutions for in vitro diagnostics kit (IVD) manufacturers. Sartorius understands the need for continuous innovation in the highly regulated and price-sensitive diagnostics market. Banczyk has expertise in medical and IVD filtration devices.
It has become the de facto standard equipment for high titer transient protein expression platforms used by many leading Chinese antibody drug pharmaceutical companies and IVD (In Vitro Diagnostics) companies since its launch in Oct. Etta Biotech is a leading cell electroporation technology and equipment supplier. JS Bio’s parent company.
Mullen As the device industry is well aware, one of the greyest areas in device regulation (of which there are many) is determining when changes to a 510(k)-cleared device trigger the need for a new clearance. By Steven J. Gonzalez & Allyson B. See 21 C.F.R. But this guidance has its limitations.
Gibbs The multi-decade battle over FDAs power to regulate Laboratory Developed Tests (LDTs) had its day in court earlier this week. Citing the May 6 compliance date for Stage 1 of the LDT Rule, both ACLA and AMP asked that Judge Jordan issue his ruling expeditiously. By Allyson B. Mullen & Jeffrey N.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

































Let's personalize your content