This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
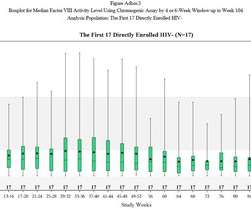
Rare diseases can often be progressive, chronic and fatal. Approximately 72 percent of rare diseases are genetic, and around 70 percent of rare geneticdiseases emerge in childhood. Sadly, one-third of children with rare diseases die before their first birthday. How Can Study Protocols Be More Effective?
Expeditious and accurate diagnoses are necessary for patients to access healthcare services and treatment options for rare geneticdiseases. Increasing the efficiency of case analysis and interpretation is essential to providing timely care for patients with geneticdiseases.
Researchers at the University of California San Francisco (UCSF) and the Whitehead Institute have developed a novel CRISPR-based tool called “CRISPRoff” that can switch off genes in human cells through epigenetic editing without altering the genetic sequence itself. It’s a great tool for controlling gene expression.”.
A 2015 study published in Nature Genetics found that the availability of human genetic data made investigational drugs twice as likely to pass pivotal trials and eventually be approved. Figure 1: The use of Mendelian randomization to validate genetic drug targets.
Researchers at the University of California San Francisco (UCSF) and the Whitehead Institute have developed a novel CRISPR-based tool called “CRISPRoff” that can switch off genes in human cells without editing the genetic sequence itself. These modifications regulate gene expression without altering the sequence or structure of DNA.
Nasdaq:RYTM), a biopharmaceutical company aimed at developing and commercializing therapies for the treatment of rare geneticdiseases of obesity, announced today that the U.S. With this approval, Imcivree becomes the first-ever FDA approved therapy for these rare geneticdiseases of obesity. BOSTON, Nov.
Novartis Gene Therapies to initiate new pivotal confirmatory study to evaluate use of AVXS-101 intrathecal (I T ) formulation in older patients with SMA to further support registration. Novartis Gene Therapies remains confident in the overall benefit-risk profile for patients on treatment.
Charlie has spent more than 26 years working on the human genome – 22 of which were spent at the Wellcome Sanger Institute, helping to assemble the human genome and trying to understand the function of the genescontained within it. We know that neurodevelopmental disorders specifically have a very high frequency of such mutations. “If
(NASDAQ: BMRN) today announced positive topline results from its ongoing global Phase 3 GENEr8-1 study of valoctocogene roxaparvovec, an investigational gene therapy for the treatment of adults with severe hemophilia A. This is the largest global Phase 3 study to date for any gene therapy in any indication, with 134 participants.
SVP, Chief Medical Officer, Novartis Gene Therapies. “We Additionally, STEER will add to the clinical data and emerging real-world evidence for the use of gene therapy to treat SMA. This route of administration has the potential to open up access for older patients to all the benefits of gene therapy.
He has pioneered the establishment of diagnostic evaluation algorithms for children with sensorineural hearing loss and developed a next generation sequencing platform to determine the genetic causes of hearing loss in children. This press release contains certain forward-looking statements concerning Sensorion and its business.
Epidiolex is the only FDA-approved formulation that contains CBD derived from the cannabis plant. CBD is the most abundant non-psychoactive cannabinoid compound found in the cannabis plant, making up about 40 percent of the plant’s extract, which contains over 100 different cannabinoid compounds.
BridgeBio is dedicated to developing therapies for geneticdiseases with unmet needs. MoCD Type A is an autosomal recessive, inborn error of metabolism caused by mutations in the molybdenum cofactor synthesis 1 gene. The approval was granted to BridgeBio Pharma, Inc. Nasdaq: BBIO ) and its affiliate Origin Biosciences, Inc.
Data will include the final analysis from the phase IIIb STASEY study of Hemlibra® (emicizumab) and updated data from the phase I/II study of SPK-8011, an AAV-based gene therapy in development by Spark Therapeutics (a member of the Roche Group). Roche’s Chief Medical Officer and Head of Global Product Development.
cTTP is a very rare, inherited and life-threatening blood clotting disorder caused by a disease-causing mutation in the ADAMTS13 (A disintegrin and metalloproteinase with thrombospondin motifs 13) gene, which encodes the ADAMTS13 enzyme that regulates blood clotting by cleaving the von Willebrand factor (VWF) protease.
Charlie has spent more than 26 years working on the human genome – 22 of which were spent at the Wellcome Sanger Institute, helping to assemble the human genome and trying to understand the function of the genescontained within it. We know that neurodevelopmental disorders specifically have a very high frequency of such mutations. “If
Targeted Augmentation of Nuclear Gene Output (TANGO) of SCN1A Reduces Seizures and Rescues Parvalbumin Positive Interneuron Firing Frequency in a Mouse Model of Dravet Syndrome. December 5, 2:00 PM – 3:30 PM; Platform A: Translational Research / Genetics. 4, 2020 14:57 UTC. BEDFORD, Mass.–( Translational Research / 2B.
PARIS–( BUSINESS WIRE )– Regulatory News: Lysogene (Paris:LYS) (FR0013233475 – LYS), a phase 3 gene therapy platform Company targeting central nervous system (CNS) diseases, announces a change in the governance and control of KGA, a company co-owned by Karen Aiach and which currently owns approximately 6% of Lysogene’s capital.
Alpha-mannosidosis is an extremely rare genetic metabolic disease affecting approximately one in 500,000 people. The lysosomal storage disorder is caused by mutations in the MAN2B1 gene, which codes for lysosomal alpha-mannosidase, an enzyme that degrades glycoproteins (proteins attached to sugar residues).
PARIS–( BUSINESS WIRE )– Regulatory News: Lysogene (FR0013233475 – LYS) (Paris:LYS), a phase 3 gene therapy platform company targeting central nervous system (CNS) diseases, today reports positive biomarker data from the ongoing AAVance clinical trial with LYS-SAF302 for the treatment of MPS IIIA (NCT03612869).
The year 2022 has proven to be a momentous period for Fulgent Genetics, marked by significant expansion of its product portfolio across various medical conditions. The company boasts an expansive portfolio of assets that grant rights to future potential royalty and milestone payments.
In the morning, I can be talking about gene therapy for hearing loss, for example, [and] in the afternoon, I can be talking about the treatment of a very specific tumour. The ABPI report also contained recommendations regarding this. So, it gives me much more breadth of exposure to be able to make that difference.”
Telomeres degrade and shorten with age and can become excessively damaged in certain geneticdiseases, as well as from lifestyle factors such as smoking, poor diet, and chronic stress. Likewise, these scientists produced “telomere-depleted” mice by inactivating a gene for telomerase, the enzyme that extends telomeres.
This condition is caused by a mutation in the fibroblast growth factor receptor 3 gene ( FGFR3 ), a negative regulator of bone growth. More than 80% of children with achondroplasia have parents of average stature and have the condition as the result of a spontaneous gene mutation. In the U.S., About BioMarin. Forward Looking Statement.
Through our in-depth understanding of the biological pathways involved in mineralization, we are pursuing the development of therapeutics to address the underlying causes of these debilitating diseases. We are initially focused on developing a novel therapy to treat the rare geneticdiseases of ENPP1 and ABCC6 deficiencies.
BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized geneticdiseases at their source. This press release contains forward-looking statements. Forward-Looking Statements.
target genes associated with cellular proliferation, angiogenesis and tumor growth. The WELIREG label contains a boxed warning that exposure to WELIREG during pregnancy can cause embryo-fetal harm. About Von Hippel-Lindau Disease. This is a rare geneticdisease with an estimated incidence of 10,000 people in the U.S.
Five patients between the ages of 5 and 15 with deletions in the maternal UBE3A gene region were enrolled in the first three cohorts and are included in the interim data analysis. Angelman syndrome is a rare, neurogenetic disorder caused by loss-of-function of the maternally inherited allele of the UBE3A gene. About Angelman Syndrome.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.































Let's personalize your content