This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This opinion from the CHMP confirms that the benefits of PAXLOVID in helping to reduce severe COVID-19-related outcomes, including hospitalization and death, in high risk patients continue to outweigh its potential risks. PAXLOVID is currently approved or authorized for conditional or emergency use in more than 70 countries to treat COVID-19.
However, if another sponsor has already obtained ODD for the same drug and condition and a marketing application has been approved, the new sponsor has the added requirement of providing a plausible hypothesis as to why the proposed drug may be clinically superior to the first already marketed drug in order to obtain ODD.
Per the FDA’s guidance on RPDD : In vitro data supporting the mechanism of action of the drug in the disease or in a related disease may suffice for rare pediatric disease designation, whereas that level of data would not generally suffice for orphan-drug designation. Criteria for Rare Pediatric Disease Designation.
However, not all LDTs currently on the market would qualify for grandfathering. FDA has, in fact, been very resistant to allowing home collection devices on the market, but that’s another story for another day.) Javitt & Jeffrey N. It has undergone dramatic recent growth, due in part to COVID-19. Emphasis added).
Eurofins Discovery will be conducting the IND-enabling in-vitro preclinical primary pharmacology and safety pharmacology studies on TD-0148A at its state-of-the-art facilities at Eurofins Cerep, DiscoverX and Panlabs. Eurofins Discovery: Wendy Parenteau, Director Marketing Communications
Email: WendyParenteau@EurofinsUS.com.
The PR first sets out to establish that it has authority to regulate in vitro diagnostic “test systems” as devices, and not just the system’s individual components, such as reagents, instruments, specimen collection devices, and software.
Statutory standards for premarket approval including de novo marketing authorization and 510(k) clearance are maintained under the program’s guidelines. Addressing this, the final guidance now contains additional information on eligibility criteria. Concerns Raised by AdvaMed. The initial STeP guidance document was drafted in 2019.
This announcement is the first traditional marketing authorization for a non-PCR based test to detect SARS-CoV-2. We are not aware of any other microbiological in vitro diagnostic test that contains the word “simple” in the regulation. symptomatic) when testing is started within 6 days of symptom onset.
Xdemvy, specifically formulated as an ectoparasiticide (anti-parasitic) for Demodex blepharitis treatment, contains the active ingredient lotilaner, a member of the isoxazoline compound family. California-based Tarsus Pharmaceuticals Inc., Xdemvy is now authorized to treat Demodex blepharitis, offering a new option for affected patients.
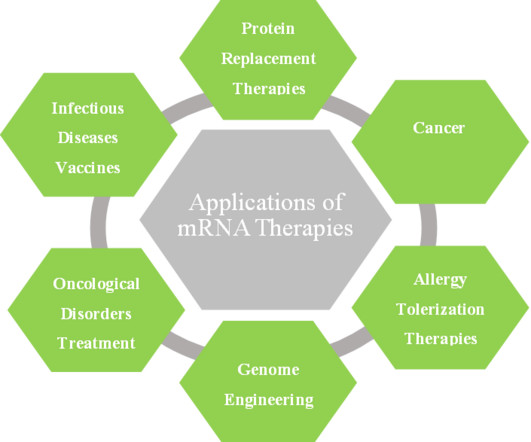
In the last few years, researchers have become interested in using in vitro transcribed (IVT) mRNA as a drug delivery agent. Given the numerous benefits of mRNA therapeutics and the increased demand for vaccines as a result of the recent COVID-19 pandemic, we believe that this market will experience significant growth in the near future.
FDA’s retrospective review of the 2003 guidance documents will include user labeling for devices that contain natural rubber and premarket approval application modular review. The 2013 guidance documents include, but are not limited to, in vitro diagnostic and clinical trial considerations.
The collaboration with Bruker announced today will assess the suitability of the test as a professional-use in-vitro diagnostic (IVD) product for SARS-CoV-2 infection to run on Bruker’s MALDI-TOF instruments for sale in the UK and Europe. I look forward to updating the market when we have definitive clinical performance data.”.
Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1, One carton contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course.
European market launch of both COVID-19 tests planned for Q1 2021.
The test is planned for commercial launch as a CE-IVD ( in vitro diagnostic) certified product in the European Union in Q1 2021.
25-minute RT-PCR lab test to complement 15-minute rapid test.
About XPhyto Therapeutics Corp. ON BEHALF OF THE BOARD.
The UK’s perceived failure to contain the initial spread of the virus was, we were told, in part due to a lesser focus on test and trace procedures than in some other countries, and that was down to us not having a tradition of diagnostics in this country. Over a year ago PCR testing was not a familiar term but is now known by general public.
and global regulatory requirements for our oral treatment, PAXLOVID™, Pfizer undertakes in vitro work (e.g., In a limited number of cases when a full virus does not contain any known gain of function mutations, such virus may be engineered to enable the assessment of antiviral activity in cells. and around the world.
Galidesivir has also demonstrated broad-spectrum activity in vitro against more than 20 RNA viruses in nine different families, including coronaviruses, filoviruses, togaviruses, bunyaviruses, arenaviruses, paramyxoviruses, and flaviviruses. The trial was conducted in Brazil under a U.S. investigational new drug application.
Coloplast is leading by example and has launched new packaging that contains no aluminum for their soft catheter SpeediCath Flex. Coloplast is leading by example and has launched new packaging that contains no aluminum for their soft catheter SpeediCath Flex. They have created a Strive25 strategy with sustainability in mind for 2025.
These clones can be manipulated and mutated in vitro to alter the expression and function of a protein. These clones can be manipulated and mutated in vitro to alter the expression and function of a protein. There are some motifs, such as hairpin loops which hinder the usability of a gene.
ADDS IN VITRO PHARMACOLOGY AND EXPANDS PROTEIN SCIENCES.
CREATES UNIQUE CO-LOCATED, FULLY-INTEGRATED, HIGH-CAPACITY R&D CENTRE.
CREATES CENTRE OF EXCELLENCE FOR STRUCTURE-BASED DRUG DESIGN.
INCREASES GLOBAL CAPACITY FOR HIGH-VALUE INTEGRATED DRUG DISCOVERY AND COMMERCIAL DEVELOPMENT OFFERING.
Dr Graham is a medicines development expert and Infectious Diseases Epidemiologist with global Biotech and Pharma R&D experience in Phase I-IV therapeutics as well as in-vivo & in-vitro diagnostics, across many modalities. THIS ANNOUNCEMENT CONTAINS INSIDE INFORMATION FOR THE PURPOSES OF ARTICLE 7 OF REGULATION (EU) NO 596/2014.
Basel, 1 September 2020 – Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that it will launch a SARS-CoV-2 Rapid Antigen Test, in late September, for markets accepting the CE Mark. Antigen test reliably and quickly triages people suspected of SARS-CoV-2, with results ready in 15 minutes, allowing informed treatment decisions.
The global GBM treatment market is projected to reach $3.3 representing the largest market.
billion by 2024, according to GlobalData, with the U.S.
The ability of a drug candidate to cross the blood brain barrier is of critical importance in treatment outcomes for CNS and brain cancers.
FDA Extends Review of Pfizer’s New Drug Application for PAXLOVID™. Tuesday, December 20, 2022 - 04:30pm. Pfizer oral treatment remains available to eligible U.S. patients under Emergency Use Authorization as a critical tool in the fight against COVID-19. NEW YORK, December 20, 2022 -- Pfizer Inc. NYSE: PFE) announced today that the U.S. In the U.S.,
Our antibody clones in their original IgG format have shown potent neutralization activity in in vitro assays and, in the case of our lead clone, in an in vivo animal model. Upon binding, the antibodies block the spike protein from interacting with ACE2 and thereby prevent virus-induced cell-killing, also known as cytopathic effect.
It is not uncommon for some in vitro diagnostic tests to suffer from poor positive predictive value, in part due to the rarity of the disease. The report was intended to make the case that FDA’s oversight of LDTs was necessary because there were rampant unvalidated and inaccurate tests on the market.
Basel, 18 September 2020 – Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced the launch of its Elecsys® Anti-SARS-CoV-2 S antibody test for markets accepting the CE Mark. The new Elecsys Anti-SARS-CoV-2 S test can quantitatively measure the level of antibodies against SARS-CoV-2 in patients who have been exposed to the virus.
Head of Medical and Marketing Division at Celltrion Healthcare. “We INCHEON, South Korea–( BUSINESS WIRE )– Celltrion Group today announced top-line results from its randomised, double-blind, and placebo controlled global Phase II/III clinical trial of CT-P59, an anti-COVID-19 monoclonal antibody treatment candidate. days; 95% C.I,
Before the FDA permits a pharmaceutical drug product to be lawfully marketed, sponsors are required to submit information about the product’s safety and efficacy so the FDA can determine : . Whether the drug’s proposed labeling (package insert) is appropriate, and what it should contain. . When Does an IND go into Effect? .
Food and Drug Administration (FDA) has designated as a Fast Track development program the investigation of Brilacidin as a potential treatment for COVID-19. Brilacidin is a first-in-class Host Defense Protein (HDP) mimetic with antiviral, anti-inflammatory and antibacterial properties.
Manufacturers must adhere to a wide array of standards and guidelines that dictate how devices are developed, tested, and brought to market. Compliance with their regulations is mandatory for manufacturers seeking to market their devices globally. Regulatory bodies such as the U.S. What is the Medical Device Safety process?
They are well established drug targets, particularly in neurological and cardiovascular diseases, but many remain undrugged or poorly drugged, and may be tractable to structure-based approaches. Metrion will contribute intellectual property, know-how and use of screening models for the nominated ion channel target. ” About Sosei Heptares.
Fosmanogepix has demonstrated broad-spectrum activity in-vitro and has shown wide distribution to various tissues including the brain, lung, kidney and eye. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time.
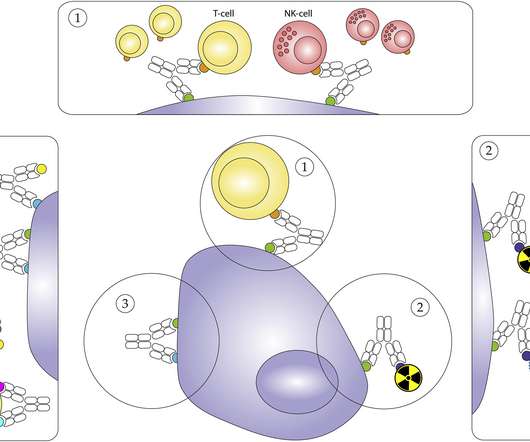
Dr Laura Moriarty, senior marketing manager at Bio-Rad, looks at the impressive immuno-therapeutic potential of bispecific antibodies (bsAbs). They are also designed to have a short in vitro half-life, meaning that they will be cleared from a patient’s system within several hours.
Progenity provides in vitro molecular tests designed to improve lives by providing actionable information that helps guide patients and physicians in making medical decisions during key life stages. SAN DIEGO, Dec. 02, 2020 (GLOBE NEWSWIRE) — Progenity, Inc. About Progenity. Progenity, Inc. Progenity, Inc. Forward Looking Statements.
CENTOGENE’s SARS-CoV-2 test is a molecular diagnostic test performed for the in vitro qualitative detection of RNA from the SARS-CoV-2 in oropharyngeal samples from presymptomatic patients and probands according to the recommended testing by public health authority guidelines.
CAMBRIDGE, Mass. and ROSTOCK, Germany and BERLIN, Nov.
(NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced real-world evidence demonstrating dramatically lower incidence rates of COVID-19 disease in individuals fully vaccinated with the Pfizer-BioNTech COVID-19 Vaccine (BNT162b2), underscoring the observed substantial public health impact of Israel’s nationwide immunization program.
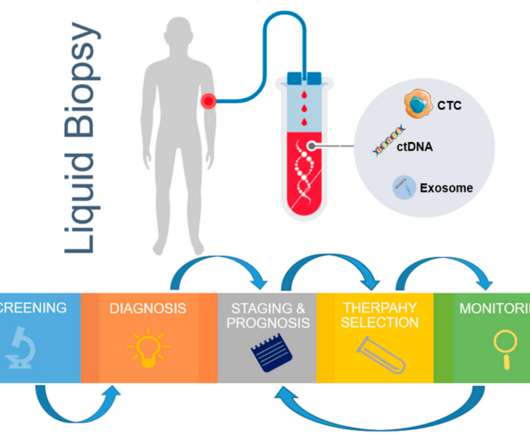
Liquid biopsy tests in oncology involve isolating entities such as circulating tumor cells (CTC), circulating tumor DNA (ctDNA) and tumor-derived exosomes. These tumor-derived entities are used to derive genomic and proteomic data. The same webinar held a Q&A session with Dr. Bahassi and two other experts from Medpace, Dr. Lyon L.
SAN DIEGO, Dec. 02, 2020 (GLOBE NEWSWIRE) — Progenity, Inc. NASDAQ: PROG) today announced the pricing of its offering of $75.0 The issuance and sale of the notes are scheduled to settle on December 7, 2020, subject to customary closing conditions. million principal amount of notes. per share of common stock.
Sulopenem has demonstrated potent in vitro activity against a wide variety of gram-negative, gram-positive and anaerobic bacteria resistant to other antibiotics. This press release contains forward-looking statements. DUBLIN, Ireland and CHICAGO, Oct. per ordinary share (or pre-funded warrant) and associated warrant.
Following the completion of the spin-off of the Upjohn Business (4) in the fourth quarter of 2020, Pfizer now operates as a focused innovative biopharmaceutical company engaged in the discovery, development, manufacturing, marketing, sales and distribution of biopharmaceutical products worldwide. per share amounts). Reported Diluted EPS (1).
(NYSE: PFE) today announced an agreement with the European Commission (EC) to supply its COVID-19 oral therapy, PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets) to countries participating in the Joint Procurement Agreement across Europe. This agreement will supply participating countries up to 3.4
.
PARTNERSHIP LEVERAGES COMPLEMENTARY STRENGTHS OF IN SILICO, IN VITRO AND IN VIVO ANTIBODY DISCOVERY IN A RANGE OF INDICATIONS.
” ABOUT ALLOY THERAPEUTICS
Alloy Therapeutics is a biotechnology company dedicated to empowering scientists in the relentless pursuit of making better medicines for all.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content