This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Although mHealth has been gaining in popularity for at least the past decade, before commercializing their mHealth products, developers must determine whether the product is subject to U.S. If so, developers must develop and execute on a regulatory strategy. The FDA’s General Approach to Regulating mHealth Products.
The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). In Part 1 of this series, general wellness devices and mobile medical applications (MMAs) were considered. Part 2 of this series is devoted to clinical decision support (CDS) software.
Shapiro — In our last post on Laboratory Developed Tests (LDTs) , we suggested that Congress, not FDA, should lead in directing modernization of LDT regulation. FDA argues that technological advances in LDTs necessitate FDA’s regulatory oversight pursuant to the Food, Drug, and Cosmetic Act (FDCA). By Jeffrey K.
You may recall that last summer (on August 19), the Department of Health and Human Services (HHS) ordered the Food and Drug Administration (FDA) to cease premarket review of laboratory developed tests (LDTs), including COVID?19 At the time, we wrote favorably about this move. Put bluntly, the review of COVID?19
Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ).
These risks have resulted in many companies being found in breach of self-regulatory codes, and a view that an increasingly narrow interpretation was being taken by authorities. What is the scope of the Guidance? Companies should ensure key words are appropriate and do not constitute unauthorised promotion to the public.
Shapiro — You may recall that last summer (on August 19), the Department of Health and Human Services (HHS) ordered the Food and Drug Administration (FDA) to cease premarket review of laboratory developed tests (LDTs), including COVID?19 At the time, we wrote favorably about this move. Put bluntly, the review of COVID?19
While this hype may be warranted in some respects—a 60-year old legal provision has now been amended to acknowledge that the science of drug development is advancing—the change is mostly symbolic and is likely to take many years before we see it have a measurable impact. Tobolowsky & Charles G. FDORA § 3209(a)(1). FDORA § 3209(a)(2).
Javitt & Philip Won — As we reported last week, FDA has issued a 26 page, single spaced, tiny-font Proposed Rule of Laboratory Developed Tests (LDTs). There is much to unpack, and we intend to do so in a series of blog posts. In this post, we focus on the proposed changes themselves, and the many questions the agency leaves unanswered.
Baumhardt provides counsel to medical device, in vitro diagnostic, and combination product manufacturers on a wide range of pre- and post-marketing regulatory topics. Baumhardt , MS, MJ, MT(ASCP), RAC, FRAPS, has joined the firm as a Senior Medical Device Regulation Expert, and that Sophia Gaulkin has joined the firm as an Associate.
On the other hand, cell free system / cell free expression system are in-vitro platforms that harness the transcription and translation machinery of living cells, in the form of cell lysates, to synthesize various types of biologics, particularly proteins. Interestingly, most of these kits are used for cell-free protein expression.
filed comments on behalf of the Coalition to Preserve LDT Access and Innovation in response to FDA’s proposed rule to regulate laboratory developed tests (LDTs) as devices. As a threshold matter, FDA lacks the power to regulate tests developed and used in a laboratory. Javitt — On Monday, Hyman, Phelps & McNamara, P.C.
Mullen — On January 18, 2024, the director of FDA’s Center for Devices and Radiological Health and the chief medical officer and acting director of CMS’ Center for Clinical Standards and Quality issued a joint press release supporting FDA’s recent proposed rule regulating Laboratory Developed Tests (LDTs).
Two weeks ago, FDA published a draft of its latest drug development guidance explaining how drug and biological product developers can use this pathway to meet the statutory standard for efficacy. the Confirmatory Evidence Guidance, available here ) had appeared on CDER’s Guidance Agenda for the past three years.
We use the term “confirmatory evidence” as short-hand for one of the two ways explicitly defined in the Federal Food Drug & Cosmetic Act (“FD&C Act”) for demonstrating substantial evidence of effectiveness. More importantly, replication is but one means to substantiate the results of a clinical investigation or scientific study.
As a result of the high healthcare cost, people from developed countries move from their home country to another to receive treatment. As a result of the high healthcare cost, people from developed countries move from their home country to another to receive treatment. What is Medical Tourism?
Javitt — For many years, one of the most controversial topics in device regulation has been the dual-track oversight of in vitro diagnostics (IVDs) and laboratory developed tests (LDTs). Enter Congress, and the introduction on June 24 of newest incarnation of the Verifying Accurate, Leading-edge IVCT Development (VALID) Act.
Cato — The long-running saga of FDA regulation of laboratory-developed tests (LDTs) has taken yet another new turn. The Notice of Proposed Rulemaking (NPRM) for LDTs is scheduled to be published in the Federal Register in August. Whether we will actually see a proposed rule in August is more than a little uncertain, however.
Overview of Cell Free Systems Cell-free systems are in-vitro platforms which allow occurrence of biochemical reactions in the absence of living cells. It is worth mentioning that the first reconstituted system, Protein Synthesis Using Recombinant Elements (PURE) , was developed in 2001.
It was developed to be administered orally so that it can be prescribed early after infection, potentially helping patients avoid severe illness (which can lead to hospitalization and death). FDA Extends Review of Pfizer’s New Drug Application for PAXLOVID™. Tuesday, December 20, 2022 - 04:30pm. NEW YORK, December 20, 2022 -- Pfizer Inc.
It was developed to be administered orally so that it can be prescribed early after infection, potentially helping patients avoid severe illness (which can lead to hospitalization and death). Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1,
Jordan ordered that FDAs Laboratory Developed Tests (LDT) Final Rule be vacated and set aside, in its entirety. In the Courts words: The final rule contemplates expanding FDAs jurisdiction to cover laboratory-developed test services as medical devices under the FDCA. Gonzalez On March 31, 2025, U.S. District Judge Sean D.
It was developed to be administered orally so that it can be prescribed early after infection, potentially helping patients avoid severe illness (which can lead to hospitalization and death). Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1
It was developed to be administered orally so that it can be prescribed early after infection, potentially helping patients avoid severe illness (which can lead to hospitalization and death). This agreement will supply participating countries up to 3.4 million treatment courses upon orders being placed.
This allows FDA to further engage with international members, address harmonization of international standards, develop multilateral agreements and cross agency documents around AI/ML and a predetermined control plan (PCCP), and converge on topics such as AI/ML. In response, Ms. This includes EUAs and full marketing authorizations.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











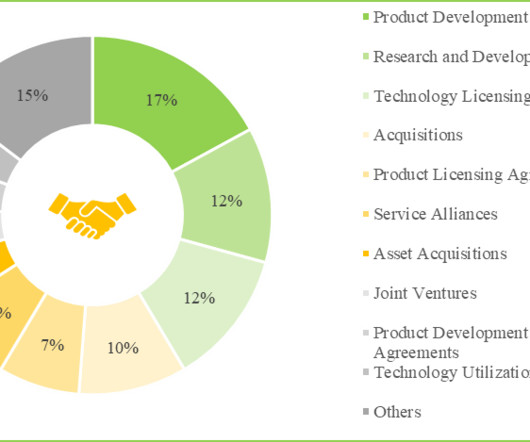












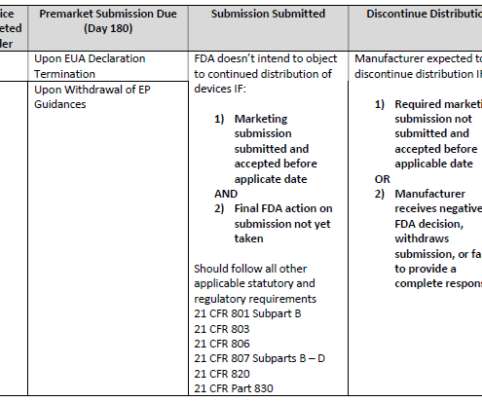





Let's personalize your content