This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The 21 st Century Cures Act generated a good deal of excitement and interest when it added a section called “ Utilizing Real World Evidence ” to the Food, Drug, and Cosmetic Act (Section 505F). The FDA recently published the ICH’s Harmonised Guideline Q13, titled “ Continuous Manufacturing of Drug Substances and Drug Products.”
The US Food and Drug Administration (FDA) has issued its first injunction under the Produce Safety Rule against Fortune Food Product Inc., The company has agreed to stop production until it takes remedial action and complies with the Federal Food, Drug, and Cosmetic Act. an Illinois-based processor of sprouts and soy products.
The first two were published in December of 2014: Providing Regulatory Submissions in Electronic Format — Submissions Under Section 745A(a) of the Federal Food, Drug, and Cosmetic Act mandates that NDAs, ANDAs, and INDs be submitted in electronic format. The FDA published three binding guidance documents regarding the TRC.
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. All packaged foods served or sold on transportation carriers (e.g., By Sophia R. Gaulkin — Last week, the U.S. 1, 2023; The applicability of food allergen labeling requirements to specific products (e.g.,
Food and Drug Administration is alerting the public that several categories of FDA-regulated products purchased from Jan. The agency is also advising that all drugs, medical devices, cosmetics and dietary supplements, regardless of packaging, be discarded. Today, the U.S. 1, 2021, through the present.
Drug manufacturers have had electronic systems in place since 2017. FDA will exercise enforcement discretion concerning the implementation of the electronic interoperable exchange of information between trading partners to the package level. Guidance at 4. So what is FDA going to do? So what is FDA going to do?
Richardson — On February 15, 2023, the Nonprescription Drugs Advisory Committee (NDAC) and the Anesthetic and Analgesic Drug Products Advisory Committee (AADPAC) held a joint meeting to discuss an application pending before FDA that would switch Narcan (naloxone) Nasal Spray from prescription to over-the-counter (OTC) status.
Farquhar — A drug manufacturer’s bad post-inspection grade from the U.S. Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDA approval of applications to market drugs manufactured at the facility. By Douglas B.
FDA Extends Review of Pfizer’s New Drug Application for PAXLOVID™. FDA Extends Review of Pfizer’s New Drug Application for PAXLOVID™. Food and Drug Administration (FDA) has extended the review period for the New Drug Application (NDA) for its COVID-19 oral treatment, PAXLOVID™ (nirmatrelvir tablets and ritonavir tablets).
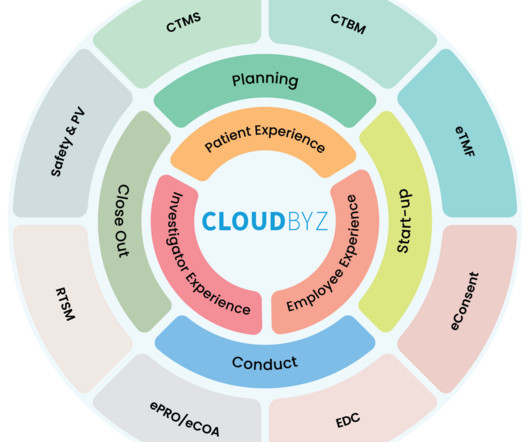
Clinical trials are the backbone of drug development, and managing these trials efficiently is paramount. Drug Supply Chain Management: Optimizes the supply chain by tracking drug inventory and distribution in real-time. In recent years, the need for a unified clinical trial management platform has become increasingly evident.
One carton contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course. Drugs listed in this section are a guide and not considered a comprehensive list of all drugs that may be contraindicated with PAXLOVID.
coli O157:H7 strain found by Michigan Department of Agriculture and Rural Development (MDARD) in Tanimura & Antle brand packaged single head romaine lettuce. brand packaged single head romaine lettuce with a pack date of Oct. recalled packaged single head romaine lettuce with a pack date of Oct. Food and Drug Administration.
The US Food and Drug Administration (FDA) defines color additives as “any substance that imparts color to a food, drug, cosmetic, or the human body. These labels must be present on the packaging of the items sold. Color additives include both synthetic substances and substances derived from natural sources.
Gaulkin assists clients across a range of pre- and post-marketing regulatory matters relating to dietary supplements, food products, drugs, medical devices, cosmetics, pet products, and consumer products. She also assists with transactional due diligence, internal investigations, and supplier subcontracts and negotiations.
2022, FDA published a draft guidance on FDA’s implementation of the Over-the-Counter Monograph Drug User Fee Program (OMUFA). FDA is authorized to charge an annual facility fee for OTC monograph drug facilities. Size, revenue, and number of finished drug products manufactured at a facility do not affect the OMUFA facility fee.
In June of 2022, Pfizer submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of PAXLOVID for patients who are at high risk of progression to severe disease from COVID-19. The target Prescription Drug User Fee Act (PDUFA) action date for a decision by the FDA is in February 2023.
After making her way from early-stage drug development research to her current role, Heather got straight to work on creating sustainable collaborations that made life better for people and their families. “I I am fascinated by the breadth of my role, which encompasses self-care products, skin health, sustainability and much more.
If the sponsor wants their drug approved, they need to complete all clinical studies and submit an application. Q: Please discuss the transfer of investigational new drug (IND) sponsorship from one sponsor to another and that process. Q: Can you ship a drug from another country to the U.S. A: Yes, the EUA is just temporary.
Computer systems used for clinical trials fall under Food and Drug Administration (FDA) 21 CFR Part 11. This applies to electronic data and signatures submitted under records requirements under other regulations such as: Federal Food, Drug, and Cosmetic Act. What is 21 CFR Part 11? Public Health Service Act.
This amendment marks the first significant revision of Part 820 since 1996, which established the Quality System (QS) regulation and “included requirements related to the methods used in, and the facilities and controls used for, designing, manufacturing, packaging, labeling, storing, installing, and servicing of devices intended for human use.”
The latest version of the bill again directs FDA to establish a standard symbol system for front-of-package labeling for conventional foods. As in the past, there are a few new things of note, and a few other things have been removed (e.g., requirements for sesame allergen labeling, which was accomplished earlier this year).
The AI response package must respond completely to all parts of the deficiencies and include all information needed to address the deficiencies; in other words, a firm cannot submit a partially completed response package and plan to submit another AI response package at a later date to address remaining deficiencies.
The Federal Food, Drug, and Cosmetic Act (FDC Act) requires that all animal foods, like human foods, be safe to eat, produced under sanitary conditions, contain no harmful substances, and be truthfully labeled. By Riëtte van Laack — FDA regulates pet food similar to other animal foods.
OTC monograph order requests or OMORs), other matters relevant to the regulation of nonprescription drugs, and the development of new nonprescription drugs under section 505G. To issue guidance that specifies the procedures and principles for formal meetings between FDA and meeting requesters for OTC monograph drugs.
To adapt ISO 13485 to the existing FDA regulatory framework, the proposed QMSR retains definitions of some terms that do not appear in ISO 13485 but are necessary to ensure alignment with the Federal Food, Drug, and Cosmetic Act (FDCA), such as the definitions for component, finished device, design validation, remanufacturer, and nonconformity.
Food and Drug Administration’s top priorities is to improve the health of Americans through better nutrition. The FDA took a critical step to help reduce sodium through a recently released guidance that establishes voluntary sodium reduction targets in processed, packaged and prepared foods. One of the U.S.
With over 2800 pharma products in 8 specific divisions that have antibiotics, cardiac, diabetes, gynec, ophthalmology, dental and cosmetic and skin products, their range of products is quite varied and impressive. They supply medications in the shape of oral syrups, tablets, powders, capsules, and drugs to the benefit of their customers.
Understanding Clinical Research in Consumer Products Companies Unlike the pharmaceutical industry, where clinical trials are inherently a part of drug discovery and approval, the consumer products industry has seen a growing emphasis on clinical research relatively recently.
115-521 ) amended Section 745(A)(b) for the Federal Food, Drug, and Cosmetic Act (FD&C Act) to include that after publication of a final guidance, pre-submissions and 510(k)s “shall be submitted solely in such electronic format as specified by the Secretary in such guidance.” Draft Guidance at 8.
As it becomes more complex with growing volumes of data, evolving regulations, and the pressure for faster drug development, traditional methods of clinical research management are no longer sufficient. The clinical research landscape is rapidly evolving. This is where the platform approach comes into play.
Pfizer’s Top 5 Best-Selling Drugs of 2022: 1) Comirnaty Comirnaty is an mRNA-based vaccine indicated for the prevention of COVID-19. Comirnaty was first approved by the US Food and Drug Administration (FDA) in August 2021 for individuals over the age of 16. billion the drug generated in 2021. billion in 2022. billion in 2022.
One carton contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course. Sufficient information is not available to assess for a potential drug interaction. Our Commitment to Access. AUTHORIZED USE. IMPORTANT SAFETY INFORMATION.
The Courts’ decisions provide insight into how natural health products are distinguished, under federal law, from cosmetics and from drugs subject to the Food and Drug Regulations. Natural health product vs. regulated drug In Winning Combination Inc. In its August 16, 2023 decision in Le-Vel Brands, LLC v.
Back in the 2017 user fee package, the FDA Reauthorization Act (also called “FDARA”), Congress set forth a process for establishing a category of OTC hearing aids—hearing aids that may be sold directly to patients without the intervention of a medical provider. Nor is it unusual for FDA to give FDA a deadline (which is often missed).
On behalf of Prescription Justice, I submitted comments to the FDA in response to its request for public comments on its draft guidance called: “Importation of Certain FDA-Approved Human Prescription Drugs, Including Biological Products, under Section 801(d)(1)(B) of the Federal Food, Drug, and Cosmetic Act: Draft Guidance for Industry.”.
Under the Federal Food, Drug, and Cosmetic Act (FDC Act) and FDAs implementing regulations, FDA has established definitions and standards of identity for certain foods. The draft guidance lays out the law, regulation, and FDAs thoughts on naming foods. For foods that do not have established definitions and standards of identity (i.e.,
Below is a copy of Gabriel Levitt’s comments on the FDA Notice of Proposed Rulemaking titled “Importation of Prescription Drugs” that were submitted Monday, March 9th, 2020. Comments on Notice of Proposed Rulemaking titled “Importation of Prescription Drugs, FDA-2019-N-5711, 84 Fed. 70796 “ Docket No.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








































Let's personalize your content