This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
On the other hand, cell free system / cell free expression system are in-vitro platforms that harness the transcription and translation machinery of living cells, in the form of cell lysates, to synthesize various types of biologics, particularly proteins. Interestingly, most of these kits are used for cell-free protein expression.
Websites – Companies can use appropriate search engine optimisation (SEO) and marketing tools to ensure that their websites are displayed high on the list of results for relevant key word searches. What is the scope of the Guidance? Companies can sponsor website content if the role of the company is made clear.
Gibbs — In 1997, Congress wisely amended the Federal Food, Drug, and Cosmetic Act (FDCA) by adding Section 513(f)(2) to establish the De Novo process. In effect, these documents serve as road signs helping to direct new market entrants. By Véronique Li, Senior Medical Device Regulation Expert & Jeffrey N.
The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). In Part 1 of this series, general wellness devices and mobile medical applications (MMAs) were considered. Part 2 of this series is devoted to clinical decision support (CDS) software.
Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ).
Baumhardt provides counsel to medical device, in vitro diagnostic, and combination product manufacturers on a wide range of pre- and post-marketing regulatory topics. In the pre-market area, Ms. In the post-market area, Ms. Prior to joining the Firm, Ms. In addition, Ms.
The crux of the proposed rule lies in the addition of ten words: “ including when the manufacturer of these products is a laboratory.” These words would be added to the definition of “ in vitro diagnostic [IVD] products” in 21 C.F.R. There is much to unpack, and we intend to do so in a series of blog posts.
Javitt — For many years, one of the most controversial topics in device regulation has been the dual-track oversight of in vitro diagnostics (IVDs) and laboratory developed tests (LDTs). The bill creates a new category, called In Vitro Clinical Tests (IVCTs), which encompasses both distributed IVD products and laboratory tests.
One would think that, as members of a great legislative body, these particular members might be at least a little concerned that FDA has been not fulfilling its “legal responsibility” under the Federal Food, Drug, and Cosmetic Act (FDCA). At the time, we wrote favorably about this move. Put bluntly, the review of COVID?19 19 LDTs again.
We’ve previously written about this proposed rule (see here , here , and here ) which would transform the diagnostic market in the United States. The Federal Food, Drug, and Cosmetic Act simply did not confer that power upon FDA. Javitt — On Monday, Hyman, Phelps & McNamara, P.C. FDA seems unconcerned by this prospect.
Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1, The additional 3.7 million treatment courses are planned for delivery by early 2023. . In June of 2022, Pfizer submitted a New Drug Application (NDA) to the U.S.
One would think that, as members of a great legislative body, these particular members might be at least a little concerned that FDA has been not fulfilling its “legal responsibility” under the Federal Food, Drug, and Cosmetic Act (FDCA). At the time, we wrote favorably about this move. Put bluntly, the review of COVID?19 19 LDTs again.
As most readers of our blog are aware, the regulatory costs to manufacturers of medical products are much lower if FDA regulates a product as a medical device rather than a drug requiring FDA marketing approval. We have blogged recently about several FDA setbacks in court ( here , for example). Add one more to that tally.
FDA Extends Review of Pfizer’s New Drug Application for PAXLOVID™. Tuesday, December 20, 2022 - 04:30pm. Pfizer oral treatment remains available to eligible U.S. patients under Emergency Use Authorization as a critical tool in the fight against COVID-19. NEW YORK, December 20, 2022 -- Pfizer Inc. NYSE: PFE) announced today that the U.S. In the U.S.,
Since 1962, the Federal Food Drug & Cosmetic Act (FDC Act) has given FDA authority to reject a drug application that fails to provide substantial evidence of effectiveness. In contrast, the new September 2023 Confirmatory Evidence Guidance is dedicated solely to the single study plus confirmatory evidence pathway.
(NYSE: PFE) today announced an agreement with the European Commission (EC) to supply its COVID-19 oral therapy, PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets) to countries participating in the Joint Procurement Agreement across Europe. This agreement will supply participating countries up to 3.4
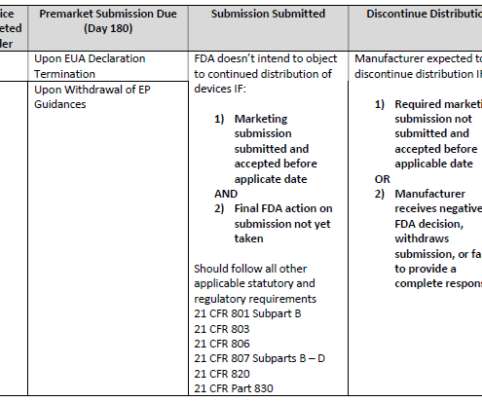
In one of the conference’s well-attended sessions, Dr. William Maisel, Chief Medical Officer and Director at CDRH’s Office of Product Evaluation and Quality, shared FDA’s experience with EUAs over the last two and a half years as well as more detail on transition planning for those devices marketed under an EUA or enforcement policy (EP).
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.





















Let's personalize your content