This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The FDA has approved Datroway (datopotamab deruxtecan-dlnk), a TROP2-directed antibody-drug conjugate (ADC) for adults with unresectable or metastatic HR-positive, HER2-negative breast cancer, developed through a global collaboration between Daiichi Sankyo and AstraZeneca. to 4,437 yen, reflecting investor confidence.
FDAApproves Oxlumo (lumasiran) for the Treatment of Primary Hyperoxaluria Type 1. Food and Drug Administration (FDA) approved Oxlumo (lumasiran) injection for subcutaneous use, the first-ever therapy available for the treatment of primary hyperoxaluria type 1 (PH1) to lower urinary oxalate levels in pediatric and adult patients.
HR+/HER2- breast cancer is the most common type of breast cancer, with the National Cancer Institute (NCI) estimating 287,850 new cases of female breast cancer in 2022 alone. What does this Approval Mean for Breast Cancer Patients? months in HR+/HER2- Patients The average five-year rate of survival is 30 percent in HR+/HER2- patients.
Paxlovid was first authorized under the FDA Emergency Use Authorization in December 2021 ; however, it has received FDAapproval on May 25, 2023. pneumoniae serotypes) replaced the company’s first pneumococcal conjugate vaccine Prevnar (PCV7, approved by the FDA in February 2000) in a February 2010 FDAapproval.
In 2023, the US Food and Drug Administration (FDA) approved a record-breaking 61 drugs, the most in history. Since January, the FDA has already signed off on more than a dozen novel drugs. The agency is reviewing applications for several highly anticipated drugs for approval in 2024.
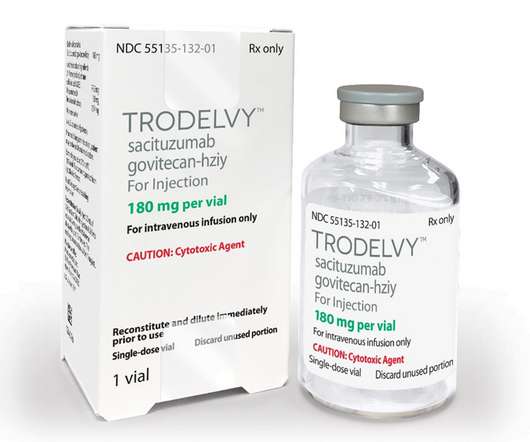
The approval is supported by data from the Phase 3 ASCENT study, in which Trodelvy demonstrated a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)), extending median PFS to 4.8 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; months vs. 6.9
The approval is based on results from the Phase 3 KEYNOTE-590 trial, which demonstrated significant improvements in overall survival (OS), progression-free survival (PFS) and objective response rate (ORR) for KEYTRUDA plus fluorouracil (FU) and cisplatin versus FU and cisplatin alone, regardless of histology or PD-L1 expression status.
today announced that the US Food and Drug Administration (FDA) has accepted the company’s supplemental Biologics License Application (sBLA) and granted Priority Review for Tecentriq® (atezolizumab) as adjuvant treatment following surgery and platinum-based chemotherapy for people with non-small cell lung cancer (NSCLC) whose tumours express PD-L1?1%,
Working with collaborators, we have conducted research where the original SARS-CoV-2 virus has been used to express the spike protein from new variants of concern. In the ongoing development of the Pfizer-BioNTech COVID-19 vaccine, Pfizer has not conducted gain of function or directed evolution research.
With nearly four years of median follow-up, data from the final analysis of the Phase 3 TITAN study confirmed that ERLEADA ® plus ADT provided a statistically significant improvement in OS with a 35 percent reduction in risk of death versus ADT alone (HR 0.65; p<0.0001). ERLEADA ® received U.S.
In the DESTINY-Gastric01 trial, patients (n=126) in the Enhertu treatment arm had a 41% reduction in the risk of death versus patients (n=62) treated with chemotherapy (based on a hazard ratio [HR] of 0.59; 95% confidence interval [CI] 0.39-0.88; 4.3] (HR=0.47; 95% CI 0.31-0.71) months [95% CI 9.6-14.3] months [95% CI 6.9-10.7]
Accelerated Approval Granted for Locally Advanced or Metastatic Urothelial Cancer Following a Platinum-Containing Chemotherapy and a PD-1/PD-L1 Inhibitor –. – New Indication Marks Second FDAApproval for Trodelvy in 2021 –. Beyond the regulatory approvals of Trodelvy in the U.S., FOSTER CITY, Calif.–(BUSINESS
The study met its primary endpoint of superior progression-free survival (PFS) as assessed by an independent review committee (IRC) with a HR 0.216 (95% CI, 0.131-0.357; p < 0.0001), demonstrating a reduction in the risk of disease progression or death for I+V of approximately 78% compared to C+O. vs. 11.4%) (p < 0.0001).
1 With up to seven years of follow-up, progression-free survival (PFS) benefit with single-agent IMBRUVICA was sustained (Hazard Ratio [HR] 0.160 [95 percent Confidence Interval (CI): 0.111–0.230]). The BTK protein sends important signals that tell B cells to mature and produce antibodies. 1 Additionally, at 6.5
one-sided), as seronegative patients treated with the antibody cocktail had a lower risk of death or receiving mechanical ventilation (HR: 0.78; 80% CI: 0.51-1.2). Casirivimab and imdevimab injection is not FDAapproved for any use. The results passed the futility analysis (p<0.3 log 10 copies/mL for combined doses).
months; hazard ratio [HR] 0.69 [95% CI, 0.58-0.83]; The HR for radiographic progression or death as assessed by blinded independent central review (BICR) was 0.864 [95% CI, 0.718–1.040]. months: HR 0.70 [95% CI, 0.60-0.83]). months: HR 0.70 [95% CI, 0.60-0.83]). 0.83]; p<0.0001). 2 ERLEADA ® received U.S.
HER2-low is characterized by the presence of some HER2 proteins on the cell surface, but not enough to be classified as HER2-positive. In the US, it is estimated that 287,850 new cases of female breast cancer will be diagnosed in 2022, according to data from the National Cancer Institute (NCI) outlined by the FDA. percent of the group.
Food and Drug Administration (FDA) approved the supplemental New Drug Application (sNDA) for Lorbrena, expanding the indication to include first-line treatment of people with anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC). The study investigated the full set of South African variant (also known as B.1.351
months with Tecentriq alone; HR=0.30, 95% CI: 0.15–0.61). TIGIT and PD-L1 are proteins that play a role in suppression of the immune system. Tiragolumab is a monoclonal antibody designed to bind with TIGIT, a protein receptor on immune cells. Tecentriq is a monoclonal antibody designed to bind with a protein called PD-L1.
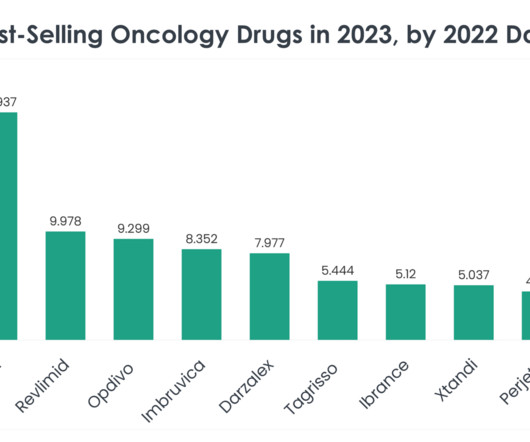
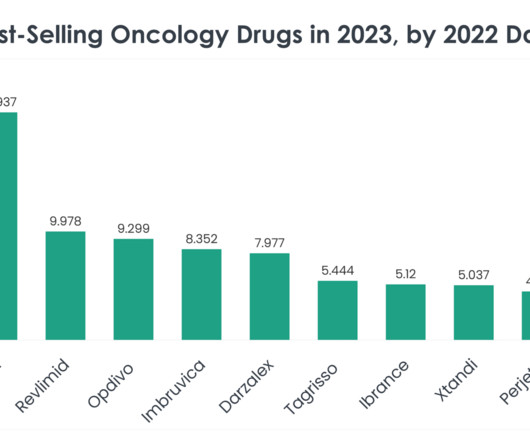
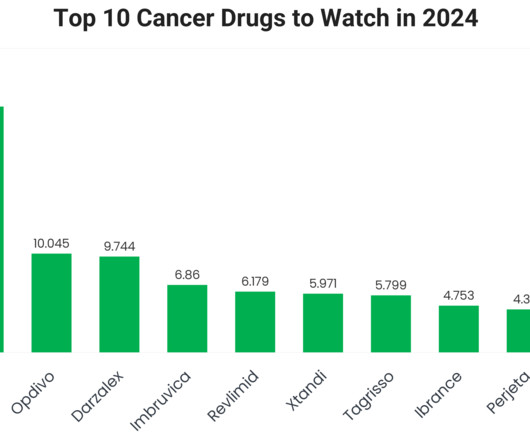
Keytruda (Pembrolizumab) Keytruda 2023 sales: $25.011 billion Company/developer: Merck Date of first US Food and Drug Administration (FDA) approval: September 4, 2014 Indications Keytruda is FDA-approved for: Unresectable or metastatic melanoma.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



















Let's personalize your content