This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The US Food and Drug Administration (FDA) has approved Alyftrek (vanzacaftor/tezacaftor/deutivacaftor), a next-in-class triple combination cystic fibrosis transmembrane conductance regulator (CFTR) modulator, to treat cystic fibrosis (CF) in patients aged six years and older with at least one F508del mutation or another responsive CFTR mutation.
Currently, the standard treatment for CAH involves glucocorticoids, which replace cortisol and help regulate hormone levels. While manageable, the condition requires lifelong treatment and monitoring to prevent complications and ensure healthy development.
A tiny child with a devastating geneticdisease who wasn’t supposed to blow out the candles on his first birthday cake. Not only did this baby survive to do all these things, but he became a poster child for gene therapy with the regulators at the U.S. One chapter discusses how to form early partnerships with regulators.
Researchers at the University of California San Francisco (UCSF) and the Whitehead Institute have developed a novel CRISPR-based tool called “CRISPRoff” that can switch off genes in human cells through epigenetic editing without altering the genetic sequence itself.
Related: FDA’s CDER Introduces New ARC Program to Accelerate Rare Disease Cures. Sanofi picked up its first Xenpozyme approval for the treatment of Niemann-Pick from Japanese regulators in March followed by an approval from European officials in May.
TSC is a rare geneticdisease that affects approximately 1 in 6,000 people. Defects in hamartin or tuberin lead to a loss of regulation of mTOR, resulting in aberrant differentiation and development, which causes the generation of enlarged cells, as seen in TSC brain lesions.
Verily, Google’s life-science-focused sibling company and Janssen will also seek to tap into the data generated by people during their everyday lives to seek for any previous health-related signals in the two years leading up to the point they consented to participate in the study as well as in the two years after.
The MeCP2 protein plays a crucial role in regulating the activity of genes involved in brain development. What’s been shown in mouse models of Rett syndrome which also have the geneticdisease, is that trofinetide helps strengthen those connections between the neurons. Daybue, also called trofinetide, is a tripeptide.
Researchers at the University of California San Francisco (UCSF) and the Whitehead Institute have developed a novel CRISPR-based tool called “CRISPRoff” that can switch off genes in human cells without editing the genetic sequence itself. These modifications regulate gene expression without altering the sequence or structure of DNA.
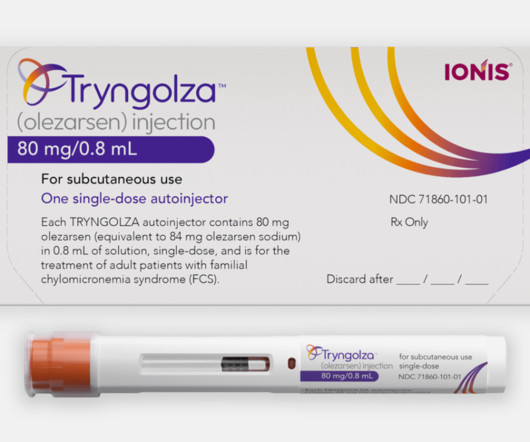
In rare disease trials, it’s not always feasible to choose clinically-relevant endpoints to measure the efficacy of a new therapeutic. Patients with this disease have a very high level of LDL-C, which increases their risk of coronary artery disease, including heart attack, stroke and atherosclerosis.
These attacks can be unpredictable, often leading to life-threatening situations when they affect the throat or lungs. HAE is caused by a deficiency or dysfunction of the C1-inhibitor, a protein involved in regulating inflammation. HAE affects approximately one in 50,000 people globally, and there is currently no cure for the disease.
cTTP is a very rare, inherited and life-threatening blood clotting disorder caused by a disease-causing mutation in the ADAMTS13 (A disintegrin and metalloproteinase with thrombospondin motifs 13) gene, which encodes the ADAMTS13 enzyme that regulates blood clotting by cleaving the von Willebrand factor (VWF) protease.
The lifesciences and healthcare are among the biggest industries globally, and their significance was particularly highlighted during the past couple of years by the COVID-19 pandemic. Given the hyperfocus on the lifesciences thanks to COVID, consumers appear to be more autonomous and vocal about their medical demands and choices.
Sooter has also found success in industry, leading and bringing together the work of different teams of researchers at NeuBase to help drive the company’s goal of developing cures and treatments for geneticdiseases, including Huntington’s Disease. Funding in the LifeScience Industry vs. Academia.
Rare diseases can often be progressive, chronic and fatal. Approximately 72 percent of rare diseases are genetic, and around 70 percent of rare geneticdiseases emerge in childhood. Sadly, one-third of children with rare diseases die before their first birthday.
FCS also severely impacts quality of life, causing chronic fatigue and recurrent stomach pain. Tryngolza works by targeting a protein in the liver, apoC-III, which regulates triglyceride metabolism. People with FCS often have triglyceride levels higher than 880 mg/dL, compared to a healthy target level of below 150 mg/dL.
The lack of existing treatments amplifies the unmet need for a viable therapy. Govorestat is the first therapy evaluated specifically for this ultra-rare condition.
Kallikrein is a serine protease enzyme that plays a key role in the kallikrein-kinin system by activating kinins, such as bradykinin, which regulate blood vessel dilation and permeability. It is currently under FDA review and, if approved, would be the first oral, on-demand HAE treatment.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.























Let's personalize your content