This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
CF is a progressive geneticdisease caused by defective CFTR proteins, which are crucial for regulating salt and water movement in cells. These mutations can vary in severity and impact on CFTR function, from complete protein dysfunction to defects in protein folding, trafficking or regulation.
Credit: UPMC PITTSBURGH, May 7, 2021 – In a paper published today in Nature Communications, an international group of collaborators led by researchers at UPMC Children’s Hospital of Pittsburgh have identified a genetic cause of a rare neurological disorder marked by developmental delay and loss of coordination, or ataxia.
MiNA Therapeutics has entered into a research collaboration and option licensing agreement with BioMarin Pharmaceutical to speed up the development of therapeutic ribonucleic acid activation (RNAa) candidates to treat rare geneticdiseases. The new deal excludes oncology and other therapeutic areas outside the scope of geneticdisease.
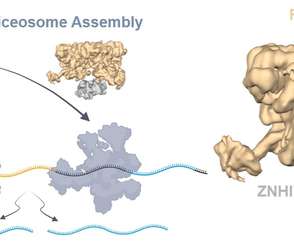
Alternative Splicing is an extraordinarily complex process that requires the coordinated action of multiple proteins, each specialised in very specific functions. These proteins are assembled and matured, forming a kind of consortium of proteins that perform these gene reading functions.
Mutations can disrupt protein binding through a “burr effect” thus interfering with the regulation of cell growth Credit: Kümmel team/Oeckinghaus team Tuberous Sclerosis Complex (TSC) affects between one and two of every 10,000 new-born babies.
Scientists illuminate the protein’s role in rare geneticdiseases often diagnosed during infancy or childhood Scientists at Scripps Research have clarified the workings of a mysterious protein called G?o, o, which is one of the most abundant proteins in the brain and, when mutated, causes severe movement disorders.
oRNA molecules have been demonstrated to possess increased stability in vivo compared to linear mRNA and can potentially create more quantities of therapeutic proteins within the body. . By self-circularisation, Orna’s oRNA technology makes circular ribonucleic acids (oRNAs) from linear RNAs.
Progress in overcoming spinocerebellar ataxia, an intractable geneticdisease Credit: Associate Professor Takahiro Seki A research team from Kumamoto University, Japan has developed an animal model that reproduces motor dysfunction and cerebellar neurodegeneration similar to that in spinocerebellar ataxia (SCA) by inhibiting chaperone-mediated autophagy (..)
Preliminary results from the study — just the second to show that CRISPR-based gene editing can be delivered systemically and performed inside the body — found the treatment, NTLA-2002, reduced levels of the disease-causing protein, kallikrein, by 65% and 92% in the low- and high-dose cohort, respectively.
Thalassaemia is a severe geneticdisease that is characterised by significantly reduced production of functional beta-globin, a component of haemoglobin, the oxygen-carrying protein in the blood. Severely-affected patients need regular blood transfusions to maintain their haemoglobin levels.
“By combining Tevard’s ability to restore the production of critical proteins with Vertex’s clinical, regulatory, and manufacturing expertise, we hope to make an important difference for patients and their families.” The company is pioneering tRNA-based therapeutics for modulating mRNA function and curing several geneticdiseases.
The Cambridge, Massachusetts biotech has discovered that areas of the non-coding parts of the human genome – referred to by its chief executive Josh Mandel-Brehm (pictured above) as the “dark side” of the genome – actually produce regRNAs that control the expression of the 2% that codes for proteins.
Nasdaq: STOK), a biotechnology company pioneering a new way to treat the underlying cause of geneticdiseases by precisely upregulating protein expression, today … Continue reading → Stoke Therapeutics Announces Proposed Public Offering Stoke Therapeutics Announces Proposed Public Offering BEDFORD, Mass.–(BUSINESS
PKU is a rare geneticdisease that manifests at birth and is marked by an inability to break down phenylalanine, an amino acid that is commonly found in many foods. So far, subjects in Phearless have received doses of either 2e13 vg/kg or 6e13 vg/kg of BMN 307, although the protocol does include a third, higher-dose group.
DEB patients are also at an increased risk of aggressive skin cancer.DEB usually presents at birth and is caused by one or more mutations in the COL7A1 gene, which encodes type VII collagen (COL7), an important protein that helps connect, strengthen and stabilize the outer and middle layers of the skin.
Seattle biotech firm Shape Therapeutics has signed a deal potentially exceeding USD 3 billion with pharma giant Roche to bolster the development of gene therapies for Alzheimer’s and Parkinson’s disease. Shape’s RNA editing technologies can modify the RNA sequence, which makes the body’s protein building blocks.
TSC is a rare geneticdisease that affects approximately 1 in 6,000 people. The disease is caused by mutations on the TSC1 and TSC2 genes, which produce the proteins hamartin and tuberin, respectively.
Premature stop codons are point mutations that disrupt protein synthesis from messenger RNA. As a consequence, patients with premature stop codon diseases have reduced or eliminated protein production from the mutation bearing allele accounting for some of the most severe phenotypes in these geneticdiseases.
Hympavzi reduces the activity of TFPI, a naturally occurring anticoagulation protein. Hympavzi is the first anti-tissue factor pathway inhibitor (anti-TFPI) approved in the US for hemophilia A or B, and it’s also the first hemophilia therapy to be administered via a pre-filled auto-injector pen.
The new drug candidate for FSHD will combine an RNA molecule from miRecule targeting double homeobox 4 (DUX4) – a protein that is mutated in FSHD – with a nanobody developed by Sanofi that targets muscle cells. billion deal in 2018.
Sarepta’s gene therapy – like rivals from Pfizer and Solid Biosciences – codes for a shortened form of the dystrophin protein that is deficient in patients with the X-linked muscle-wasting disease, which occurs primarily in males.
CRISPR drugs can be used to modify the expression of disease-associated proteins in the body, for example, by correcting a mutation in a specific gene. There is another $1 billion in downstream payments on offer, assuming the drug candidates all make it through to regulatory approval and hit sales targets.
Through its Shielded Living Therapeutics platform, the company is developing functional cures for chronic diseases. The impact of CNS diseases extends beyond patients—to their families and society as well.” Looking to extend the human healthspan, BioAge raised $90 million in an oversubscribed Series C.
Protein Based Assays- These tests measure the presence and levels of specific proteins, which can serve as biomarkers for various diseases. These tests can aid in the diagnosis and treatment monitoring of various conditions by providing information about the underlying biological processes and disease states.
HAE is caused by a deficiency or dysfunction of the C1-inhibitor, a protein involved in regulating inflammation. HAE affects approximately one in 50,000 people globally, and there is currently no cure for the disease. These attacks can be unpredictable, often leading to life-threatening situations when they affect the throat or lungs.
Dietary changes, including restriction of salt and animal protein, are also recommended. Symptoms and Etiology: Characterized by progressive muscle weakness and atrophy, Duchenne muscular dystrophy (DMD) is an X-linked genetic condition that primarily affects males.
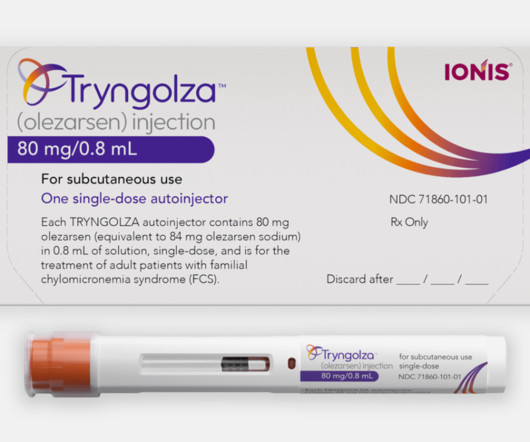
Tryngolza works by targeting a protein in the liver, apoC-III, which regulates triglyceride metabolism. People with FCS often have triglyceride levels higher than 880 mg/dL, compared to a healthy target level of below 150 mg/dL. FCS also severely impacts quality of life, causing chronic fatigue and recurrent stomach pain.
The MeCP2 protein plays a crucial role in regulating the activity of genes involved in brain development. What’s been shown in mouse models of Rett syndrome which also have the geneticdisease, is that trofinetide helps strengthen those connections between the neurons. Daybue, also called trofinetide, is a tripeptide.
Haemophilia A is a serious, inherited bleeding disorder in which a person’s blood doesn’t clot properly, as they either lack or do not have enough of a clotting protein called factor VIII. 11 People with haemophilia A either lack or do not have enough of a clotting protein called factor VIII.
Miplyffa primarily slows disease progression using a different mechanism of action, targeting heat shock protein responses to assist cells under stress.
AavantiBio’s strategic partnership with University of Florida’s Powell Gene Therapy Center provide their foundational research in rare genetic disorders. The company’s lead program is aimed at Friedrich’s Ataxia, a rare inherited geneticdisease that causes cardiac and central nervous system dysfunction. Be Biopharma .
Apitegromab is a monoclonal antibody that selectively binds to and inhibits myostatin, a protein that limits muscle growth. Current therapies mainly address motor neuron loss, leaving a need for treatments that directly target muscle weakness.
Telomeres degrade and shorten with age and can become excessively damaged in certain geneticdiseases, as well as from lifestyle factors such as smoking, poor diet, and chronic stress. Shortening of telomeres is associated with the symptoms of aging, heart disease, DNA damage and uncontrolled cell replication, which can lead to cancer.
The US Food and Drug Administration (FDA) has approved Takeda Pharmaceuticals’ Adzynma, the first recombinant protein product for prophylactic (preventive) or on‑demand enzyme replacement therapy (ERT) in adult and pediatric patients with congenital thrombotic thrombocytopenic purpura (cTTP), an ultra-rare blood clotting disorder.
Alpha-mannosidosis is an extremely rare genetic metabolic disease affecting approximately one in 500,000 people. The lysosomal storage disorder is caused by mutations in the MAN2B1 gene, which codes for lysosomal alpha-mannosidase, an enzyme that degrades glycoproteins (proteins attached to sugar residues).
This progressive disease occurs when misfolded transthyretin (TTR) proteins form amyloid deposits in the heart, leading to cardiac dysfunction. BridgeBio has been highly active in the geneticdiseases space.
The interim analysis did not include data from other exploratory outcome measures such as seizure frequency, sleep diaries, EEG patterns, UBE3A protein levels in the CSF, ambulation by wearable device, and adaptive behaviors. This condition is typically not inherited but instead occurs spontaneously. About Ultragenyx.
The most common adverse events associated with valoctocogene roxaparvovec occurred early and included transient infusion-associated reactions and transient, asymptomatic, and mild to moderate rise in the levels of certain proteins and enzymes measured in liver function tests with no long-lasting clinical sequelae. GENEr8-1 Study Description.
Research and development in the area is currently growing at a fast rate, and the National Institute of Health reports hundreds of clinical trials to test gene therapies for different geneticdiseases, immune system disorders, oncology treatments, neurogenerative diseases, infectious diseases, and more.
Through RNA silencing, it targets the expression of antithrombin, a protein that inhibits blood clotting. Moreover, Qfitlia, an siRNA therapy that blocks target protein expression at the RNA level, requires dosing only once every other month, compared to Alhemos daily and Hympavzis weekly dosing.
CALD is caused by mutations in the ABCD1 gene located on the X chromosome, which provides instructions for the production of the ALD protein. ALD protein is needed to clear toxic molecules called very long-chain fatty acids (VLCFAs) in the brain, and if mutated causes the VLCFAs to accumulate and damage the myelin sheath.
With the rapid development of biotechnology and molecular medicine, the introduction of mRNA as a vaccine or therapeutic agent enables the production of almost any desired functional protein/peptide within the human body.
Stoke believes that STK-001, a proprietary antisense oligonucleotide (ASO), has the potential to be the first disease-modifying therapy to address the genetic cause of Dravet syndrome. protein expression by leveraging the non-mutant (wild-type) copy of the SCN1A gene to restore physiological Na V 1.1
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








































Let's personalize your content