This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The Central Drugs Standard Control Organisation (CDSCO) and Indian Council of Medical Research (ICMR) have together released the draft standard evaluation protocols for the purpose of issuing license for in-vitro diagnostics (IVDs) under the Medical Devices Rules (MDR), 2017.
The Central Drugs Standard Control Organisation (CDSCO) has requested stakeholders to take appropriate action for timely reporting of adverse events related to medical devices to the Materiovigilance Programme of India (MvPI), in the backdrop of shifting the segment to licensing regime.
In the last three years alone, there have been over 633,000 patents filed and granted in the pharmaceutical industry, according to GlobalData’s report on Immuno-oncology in Pharmaceuticals: In-vitro T-cell activation. Immatics is the leading patent filer in in-vitro T-cell activation.
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
The FDA’s General Approach to Regulating mHealth Products. Food and Drug Administration (FDA) regulation as a medical device. Determining whether a product is a medical device subject to FDA regulation necessarily begins with understanding the FDA regulatory definition of ‘medical device’. not a medical device, ii.
Bourla added that, “At the same time, we are making the right investments and engaging in the appropriate conversations with regulators to help position us to potentially develop and seek authorization for an updated mRNA vaccine or booster if needed.”. The third booster will be given to participants from the Phase I study in the US.
Criteria for regulation. The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). peer-reviewed clinical studies or clinical practice guidelines) meet Criterion 2 and are not medical devices subject to FDA regulation and oversight.
1] Yet FDA’s conclusions about the Agency’s ability to regulate the entire laboratory industry are based on fundamentally flawed assumptions about the number of entities and tests that will be subject to FDA regulation. 5] This stratification, though, assumes that LDTs will follow the same pattern as IVDs currently regulated by FDA.
Eurofins Discovery will be conducting the IND-enabling in-vitro preclinical primary pharmacology and safety pharmacology studies on TD-0148A at its state-of-the-art facilities at Eurofins Cerep, DiscoverX and Panlabs. FDA Investigational New Drug (“IND”)-enabling pharmacology studies. About BetterLife Pharma: BetterLife Pharma Inc.
Dr Graham is a medicines development expert and Infectious Diseases Epidemiologist with global Biotech and Pharma R&D experience in Phase I-IV therapeutics as well as in-vivo & in-vitro diagnostics, across many modalities. He has in depth Global Development Expertise (e.g. Previously, he held roles as CMO at Trimeris Inc.
Food and Drug Administration (FDA) has designated as a Fast Track development program the investigation of Brilacidin as a potential treatment for COVID-19. Brilacidin is a first-in-class Host Defense Protein (HDP) mimetic with antiviral, anti-inflammatory and antibacterial properties.
We plan to submit these data to FDA as a proposed amendment to our Emergency Use Authorization in the coming weeks and to other regulators around the world, with the hope of starting to vaccinate this age group before the start of the next school year.”. How Safe and Effective is Pfizer’s COVID-19 Vaccine for Kids?
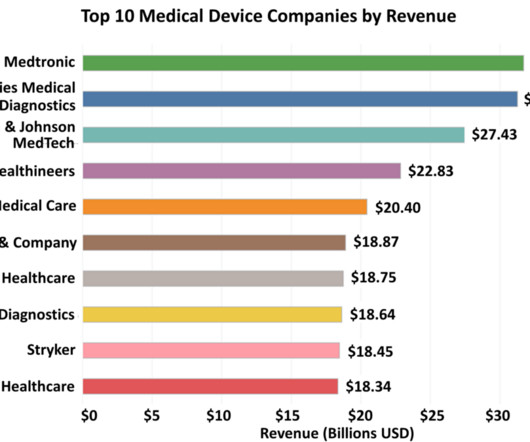
As technology continues to revolutionize every sector of our lives, the medical device industry stands at the forefront of this innovation, playing a pivotal role in enhancing patient care, improving diagnostic accuracy and transforming treatment modalities. Note: When it comes to companies that report in foreign currencies, the conversion to U.S.
Thirty years ago today, FDA announced that it had the authority to regulate you. Food and Drug Administration, Compliance Policy Guide, Commercialization of Unapproved In Vitro Diagnostic Devices Labeled for Research and Investigation (Aug. By Jeffrey N. Gibbs & Allyson B. Mullen — Happy Birthday Laboratory Developed Tests (LDTs).
Introduction to Medical Device Safety, Systems, and Regulations The medical device industry is a rapidly evolving field that plays a critical role in modern healthcare. This is where the importance of stringent safety processes, robust systems, and comprehensive regulations comes into play. What is the Medical Device Safety process?
OHMX.bio – Belgium-based OHMX.bio and Fujirebio Europe were awarded a €720,000 research grant from Flanders Innovation & Entrepreneurship (VLAIO) to develop a clinical in vitro diagnostics (IVD) platform incorporating third generation sequencing (TGS) technologies. A solution could be at hand with Overland Pharmaceuticals.
The European Commission (EC) will review the CHMP recommendation and is soon expected to make a final decision. This opinion from the CHMP confirms that the benefits of PAXLOVID in helping to reduce severe COVID-19-related outcomes, including hospitalization and death, in high risk patients continue to outweigh its potential risks.
FDA Extends Review of Pfizer’s New Drug Application for PAXLOVID™. Tuesday, December 20, 2022 - 04:30pm. Pfizer oral treatment remains available to eligible U.S. patients under Emergency Use Authorization as a critical tool in the fight against COVID-19. NEW YORK, December 20, 2022 -- Pfizer Inc. NYSE: PFE) announced today that the U.S. In the U.S.,
Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1, The additional 3.7 million treatment courses are planned for delivery by early 2023. . In June of 2022, Pfizer submitted a New Drug Application (NDA) to the U.S.
The antibody combination was granted a Special Approval Pathway under article 14-3 of the Pharmaceuticals and Medical Devices Act. “Ronapreve has been shown to improve survival in high-risk, non-hospitalised COVID-19 patients by reducing the risk of hospitalisation and death. About Ronapreve (casirivimab and imdevimab).
Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1 I am grateful to have received four doses of the Pfizer-BioNTech vaccine and I am feeling well while experiencing very mild symptoms. FDA Emergency Use Authorization Statement.
(NYSE: PFE) today announced an agreement with the European Commission (EC) to supply its COVID-19 oral therapy, PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets) to countries participating in the Joint Procurement Agreement across Europe. This agreement will supply participating countries up to 3.4
and global regulatory requirements for our oral treatment, PAXLOVID™, Pfizer undertakes in vitro work (e.g., and global regulators for all antiviral products and are carried out by many companies and academic institutions in the U.S. It is important to note that these studies are required by U.S. and around the world.
Raises Full-Year 2021 Guidance (3) for Revenues to a Range of $70.5 Billion and Adjusted Diluted EPS (2) to a Range of $3.55 Now Anticipates Revenues of Approximately $26 Billion for BNT162b2, Reflecting 1.6 Billion Doses Expected to be Delivered in 2021 Under Signed Contracts as of Mid-April 2021. per share amounts). Reported Diluted EPS (1).
Data from the registrational COMET-ICE trial also will form the basis for a Biologics License Application (BLA) submission to the FDA. GSK and Vir will continue discussions with the European Medicines Agency (EMA) and other global regulators to make VIR-7831 available to patients with COVID-19 as soon as possible. About COMET-ICE.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.




























Let's personalize your content