This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Shapiro — In our last post on Laboratory Developed Tests (LDTs) , we suggested that Congress, not FDA, should lead in directing modernization of LDT regulation. This step would help propel forward a public conversation (which CMS has already started ) about how to update these regulations in light of technological advances.
TriClip G4 System Manufacturer/developer : Abbott Medical Date of FDA approval : April 1, 2024 Approved for : Tricuspid regurgitation (TR). The tricuspid valve, one of the heart’s four valves, regulates blood flow from the right atrium to the right ventricle, preventing backflow between these chambers.
is pleased to announce that Director Allyson Mullen has been appointed to the Board of Directors of the Association of Medical Diagnostic Manufacturers (AMDM). Mullen’s extensive experience and contributions to the field of in vitro diagnostic (IVD) regulation. This prestigious appointment recognizes Ms.
Thirty years ago today, FDA announced that it had the authority to regulate you. Food and Drug Administration, Compliance Policy Guide, Commercialization of Unapproved In Vitro Diagnostic Devices Labeled for Research and Investigation (Aug. By Jeffrey N. Gibbs & Allyson B. Mullen — Happy Birthday Laboratory Developed Tests (LDTs).
Gibbs — On March 21, 2024, the House Energy and Commerce held a subcommittee hearing titled “Evaluating Approaches to Diagnostic Test Regulation and the Impact of the FDA’s Proposed Rule.” FDA, which was not invited to participate, would surely have concurred. This seems to have been the outcome that many lawmakers desired.
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
The filing with the FDA follows approval by several regulators across the world, including in the European Union and Japan. Originally intended as a treatment for Ebola virus, Veklury is a nucleotide analogue with broad-spectrum antiviral activity both in vitro and in vivo in animal models against several emerging viruses.
XTALKS WEBINAR: Impacts of the New IVD Regulation (IVDR) for Manufacturers and Users Live and On-Demand: Tuesday, January 9, 2024, at 10am EST (4pm CET/EU-Central) Register for this free webinar to learn how the new In Vitro Diagnostic Regulation (IVDR) impacts manufacturers and users of diagnostic devices.
Today, FDA released a copy of a proposed rule to regulate laboratory-developed tests (LDTs), which is scheduled to be published in the Federal Register on October 3rd. For more than 30 years, FDA has asserted that it has jurisdiction to regulate LDTs as medical devices and clinical laboratories as manufacturers.
Baumhardt, Senior Medical Device Regulation Expert & Richard A. The program began accepting proposals on September 7, 2022, from manufacturers that have existing technologies and can scale production, while meeting FDA quality requirements.
The guidance comes from the ACCESS Consortium of regulators from the UK, Australia, Canada, Singapore and Switzerland. ” BIVDA, the trade association for British in-vitro diagnostic companies, supported the “super-deduction” policy. .”
Baumhardt , MS, MJ, MT(ASCP), RAC, FRAPS, has joined the firm as a Senior Medical Device Regulation Expert, and that Sophia Gaulkin has joined the firm as an Associate. Baumhardt provides counsel to medical device, in vitro diagnostic, and combination product manufacturers on a wide range of pre- and post-marketing regulatory topics.
FDA , the Court of Appeals ruled that FDA cannot regulate a medical product – in this case, the radiographic contrast agent barium sulfate – as a drug when the product meets the definition of a device. The decision has wide-ranging implications for FDA’s assertion of discretion in classifying and regulating medical products.
A proposal has been put forward to amend the transitional provisions for certain medical devices and in vitro diagnostic medical devices (amending Regulations (EU) 2017/745 (MDR) 1 and (EU) 2017/746 (IVDR) 2. – The extension is directly applicable, therefore changing the date on the individual certificates is not necessary.
Loloei tackled legal matters related to various aspects of the regulation of medical devices, in vitro diagnostics, and combination products including regulatory and compliance issues, dispute resolutions between the FDA and sponsors, and FDA enforcement actions. While at FDA, Ms. During her FDA tenure, Ms. Gibbs , HP&M Director.
Laboratories struggling to understand the myriad implications of being regulated as device “manufacturers” were hopeful that additional guidance would shed light on how to apply FDA’s existing medical device regulatory framework to their operations. 803), Reporting of Corrections and Removals (21 C.F.R. §
The crux of the proposed rule lies in the addition of ten words: “ including when the manufacturer of these products is a laboratory.” These words would be added to the definition of “ in vitro diagnostic [IVD] products” in 21 C.F.R. There is much to unpack, and we intend to do so in a series of blog posts.
We’ve got so many great life science assets in the UK, but there are still some gaps – manufacturing capacity being one of them.”. “We There have also been many pragmatic collaborations between companies, sites and regulators that have helped keep research and HTA going – such as the more flexible rules introduced by the MHRA.”.
Complaints FDA discussed how LDT developers should handle the transition from the current Quality System Regulation (QSR)(21 CFR Part 820) to the recently promulgated Quality Management System Regulation (QMSR) that is scheduled to take effect on February 2, 2026. By Steven J. Gonzalez & Lisa M.
CerTest Biotec specializes in the development and manufacturing of in vitro diagnostic medical devices and has expertise in the detection of human diseases. In a joint press release , the companies said the aim of the partnership is to design a diagnostic test that can help advance understanding of the global spread of the disease.
Gibbs — It is widely expected that the fate of the VALID Act – and therefore the world of diagnostic regulation – will be determined in the next two weeks (see our previous post here ). Javitt & Jeffrey N. This is not a trivial matter. Telehealth has become increasing important to the health care system.
Mullen — FDA has been clearing over-the-counter (OTC) in vitro diagnostic (IVD) tests nearly since the beginning of its premarket regulation of devices. The first OTC IVD cleared by FDA was a qualitative dipstick urine glucose test in 1977, followed shortly thereafter by the first OTC pregnancy test clearance in 1978.
1] Yet FDA’s conclusions about the Agency’s ability to regulate the entire laboratory industry are based on fundamentally flawed assumptions about the number of entities and tests that will be subject to FDA regulation. 5] This stratification, though, assumes that LDTs will follow the same pattern as IVDs currently regulated by FDA.
These risks have resulted in many companies being found in breach of self-regulatory codes, and a view that an increasingly narrow interpretation was being taken by authorities. What is the scope of the Guidance? Information on social media and digital channels should be kept up to date and date stamped, with the date posted or last updated.
Introduction to Medical Device Safety, Systems, and Regulations The medical device industry is a rapidly evolving field that plays a critical role in modern healthcare. This is where the importance of stringent safety processes, robust systems, and comprehensive regulations comes into play. What is the Medical Device Safety process?
Sartorius is a globally recognized player in the diagnostics industry, providing a variety of solutions for in vitro diagnostics kit (IVD) manufacturers. Sartorius understands the need for continuous innovation in the highly regulated and price-sensitive diagnostics market. Xu joined Sartorius in 2002.
In vitro diagnostic (IVD) devices are tests used on human biospecimens (e.g., Additionally, while the Food and Drug Administration (FDA) regulates all IVDs as medical devices, the agency has generally not enforced device regulations for certain IVDs designed, manufactured, and used within a single CLIA-certified laboratory.
We share the public’s concerns regarding the new and continued safety issues of CPAP machines and certain recalled medical devices manufactured by Philips. Addressing these safety concerns remains a top priority for the FDA.” It is used at home and in clinical settings. In case any issue is detected, the machine should not be used.
Vittoria’s investigational CAR-T therapy leverages gene editing to modulate the CD5 signaling pathway and a shorter manufacturing time to enhance efficacy and improve manufacturing efficiencies and vein-to-vein time. Such challenges highlight a significant unmet need and call for further advancements in the field.
Whether the methods used in manufacturing the drug and the controls used to maintain the drug’s quality are adequate to preserve the drug’s identity, strength, quality, and purity.” . The Food and Drug Administration (FDA) is the federal entity in the U.S. When Does an IND go into Effect? . The IND goes into effect; and .
In response to a growing demand for faster, approved, and cost-effective medical devices for chronic diseases, medical device manufacturers and emerging biotech companies are facing increasingly complex pathways to successfully bring their innovative ideas to the market. Meeting with the Regulators. Regulators are not your adversaries.
Unlike March 2020, numerous tests by multiple manufacturers have been reviewed by FDA and are being distributed. This article ( link ), which is made available with the permission of the Food and Drug Law Institute, examines multiple facets of FDA’s regulation of COVID assays. The onset of COVID brought unprecedented challenges to FDA.
Manufacturers regularly review the design of their product, including labelling and IFU based on customer feedback. In the early phases of the COVID-19 pandemic, in vitro diagnostics (IVDs) were rapidly developed, validated and verified, and then rolled out. Check the IFU for each incoming consignment to detect any changes to the IFU.
In particular, these members of Congress seem to be requesting that HHS revive a dysfunctional (and probably unconstitutional ) approach to regulation of LDTs. Given this stated concern about test accuracy and validation, why are they only urging FDA to regulate COVID?19 At the time, we wrote favorably about this move. 19 LDTs again.
Baumhardt, Senior Medical Device Regulation Expert & Richard A. Lewis, Senior Regulatory Device & Biologics Expert — We are almost three years into the public health emergency as a result of the COVID-19 pandemic, and still only have 19 rapid antigen tests authorized for at-home use in the United States. That could happen again.
Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1, The additional 3.7 million treatment courses are planned for delivery by early 2023. . In June of 2022, Pfizer submitted a New Drug Application (NDA) to the U.S.
Unlike March 2020, numerous tests by multiple manufacturers have been reviewed by FDA and are being distributed. This article ( link ), which is made available with the permission of the Food and Drug Law Institute, examines multiple facets of FDA’s regulation of COVID assays. The onset of COVID brought unprecedented challenges to FDA.
With the increased interest and gradual shift of investment from small molecule drugs to biologics and the establishment of several biologics manufacturing companies / biologics CMOs, more than 250 biologic therapies and vaccines have been developed, globally. Like all drugs, biologics are regulated by the FDA.
and global regulatory requirements for our oral treatment, PAXLOVID™, Pfizer undertakes in vitro work (e.g., and global regulators for all antiviral products and are carried out by many companies and academic institutions in the U.S. It is important to note that these studies are required by U.S. and around the world.
Other articles examined communication – one on persuasive skills, another on disseminating of regulatory intelligence – and the regulatory response to nitrosamine contamination of drug products. . . Continuous professional development. DiMichele provides valuable information on some of the available programs. . .
The companies will collaborate on developing and manufacturing REGN-COV2. We’re committing our manufacturing expertise and capacity, and our global distribution network to bring Regeneron’s potential antibody combination to as many people around the world as we possibly can.”. Regeneron will distribute REGN-COV2 in the U.S.
In particular, these members of Congress seem to be requesting that HHS revive a dysfunctional (and probably unconstitutional ) approach to regulation of LDTs. Given this stated concern about test accuracy and validation, why are they only urging FDA to regulate COVID?19 At the time, we wrote favorably about this move. 19 LDTs again.
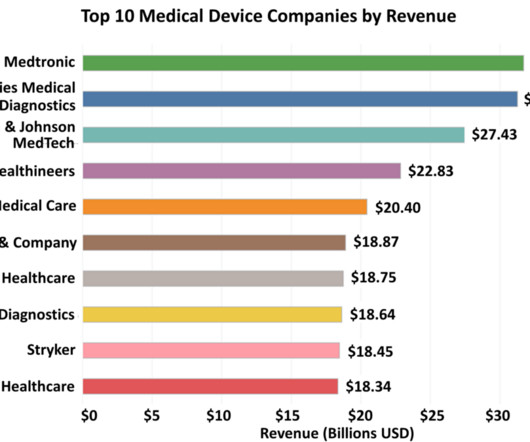
Medtronic, the leading global medical device manufacturer, reported a revenue growth of 5 percent in the last fiscal year, affirming its position as the top global medical technology company by revenue. This article takes a comprehensive look at the top 10 medical device companies that are leading the charge in this rapidly evolving field.
The European Commission (EC) will review the CHMP recommendation and is soon expected to make a final decision. This opinion from the CHMP confirms that the benefits of PAXLOVID in helping to reduce severe COVID-19-related outcomes, including hospitalization and death, in high risk patients continue to outweigh its potential risks.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content