This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
In the last three years alone, there have been over 633,000 patents filed and granted in the pharmaceutical industry, according to GlobalData’s report on Immuno-oncology in Pharmaceuticals: In-vitro T-cell activation. Immatics is the leading patent filer in in-vitro T-cell activation.
Despite entering its fourth decade of availability, in vitro fertilization (IVF), a medical technique used to facilitate the conception of a baby for those facing fertility problems, remains an elusive dream for many. Fairtility also claims the product reduces the time taken for each IVF cycle by 30 hours. in 2000 to 12.3%
The Camargo Blog is publishing a four-part blog series highlighting those designation programs available specifically for products with rare disease indications: Orphan Drug Designation (ODD), Rare Pediatric Disease Designation (RPDD), and Humanitarian Use Device (HUD) designation. Definition of a Rare Disease or Condition.
The Camargo Blog is publishing a four-part blog series highlighting those designation programs available specifically for products with rare disease indications : Orphan Drug Designation (ODD), Rare Pediatric Disease Designation (RPDD), and Humanitarian Use Device (HUD) designation.
Thirty years ago today, FDA announced that it had the authority to regulate you. Not yet understanding how important you’d become, you entered the regulatory world without a name – the Agency simply referred to you as “home brew” products. Mullen — Happy Birthday Laboratory Developed Tests (LDTs). 92P-0405 (Aug.
Gibbs — On March 21, 2024, the House Energy and Commerce held a subcommittee hearing titled “Evaluating Approaches to Diagnostic Test Regulation and the Impact of the FDA’s Proposed Rule.” FDA, which was not invited to participate, would surely have concurred. This seems to have been the outcome that many lawmakers desired.
The FDA’s General Approach to Regulating mHealth Products. Although mHealth has been gaining in popularity for at least the past decade, before commercializing their mHealth products, developers must determine whether the product is subject to U.S. Food and Drug Administration (FDA) regulation as a medical device.
Unlike CAR T therapies, TILs are produced in vitro by purifying natural infiltrating lymphocytes from the patient’s own tumor microenvironment. Its primary challenges include: Production Delays : The time-intensive process of expanding TILs to therapeutic quantities poses logistical hurdles.
Javitt — For many years, one of the most controversial topics in device regulation has been the dual-track oversight of in vitro diagnostics (IVDs) and laboratory developed tests (LDTs). At various points and in various ways, FDA has sought – mostly unsuccessfully – to regulate LDTs (see here ).
Baumhardt, Senior Medical Device Regulation Expert & Adrienne R. Lenz, Principal Medical Device Regulation Expert — FDA recently released a new eSTAR template for device pre-submissions and 513(g) Requests for Information, referred to as PreSTAR. package labeling is optional).
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
By Véronique Li, Senior Medical Device Regulation Expert — A joint eSTAR pilot (which we previewed in November) between FDA and Health Canada has now been launched. Nine participants will be selected to use the non-In Vitro Diagnostic eSTAR. checks for incomplete sections. checks for incomplete sections.
Mullen — On January 31, 2024, FDA announced its intent to initiate the reclassification process for most in vitro diagnostic (IVD) products that are currently class III (high risk) into class II (moderate risk). By Steven J. Gonzalez & Allyson B. First, all this would do is move the premarket submission from one bucket to another.
By Véronique Li, Senior Medical Device Regulation Expert & Philip Won — Last week, our blog post advised planning a transition strategy in advance of the news of the termination of the Covid-19 public health emergency. The 2013 guidance documents include, but are not limited to, in vitro diagnostic and clinical trial considerations.
Baumhardt , MS, MJ, MT(ASCP), RAC, FRAPS, has joined the firm as a Senior Medical Device Regulation Expert, and that Sophia Gaulkin has joined the firm as an Associate. Baumhardt provides counsel to medical device, in vitro diagnostic, and combination product manufacturers on a wide range of pre- and post-marketing regulatory topics.
FDA , the Court of Appeals ruled that FDA cannot regulate a medical product – in this case, the radiographic contrast agent barium sulfate – as a drug when the product meets the definition of a device. The decision has wide-ranging implications for FDA’s assertion of discretion in classifying and regulating medical products.
Loloei tackled legal matters related to various aspects of the regulation of medical devices, in vitro diagnostics, and combination products including regulatory and compliance issues, dispute resolutions between the FDA and sponsors, and FDA enforcement actions. While at FDA, Ms. During her FDA tenure, Ms.
IS: Our genotoxicity testing includes the core battery of the tests requested by EMEA/ICH including mutagenicity in vitro, chromosomal aberration test in vitro and micronucleus test in vivo. Natalie Coomber: Can you provide a quick background on the history of Biotest and the main work it is currently undertaking?
Criteria for regulation. The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). peer-reviewed clinical studies or clinical practice guidelines) meet Criterion 2 and are not medical devices subject to FDA regulation and oversight.
The crux of the proposed rule lies in the addition of ten words: “ including when the manufacturer of these products is a laboratory.” These words would be added to the definition of “ in vitro diagnostic [IVD] products” in 21 C.F.R. There is much to unpack, and we intend to do so in a series of blog posts.
1] Yet FDA’s conclusions about the Agency’s ability to regulate the entire laboratory industry are based on fundamentally flawed assumptions about the number of entities and tests that will be subject to FDA regulation. 5] This stratification, though, assumes that LDTs will follow the same pattern as IVDs currently regulated by FDA.
Baumhardt, Senior Medical Device Regulation Expert & Richard A. The program began accepting proposals on September 7, 2022, from manufacturers that have existing technologies and can scale production, while meeting FDA quality requirements.
Type I interferons are critical molecular immune regulators that are at the forefront of antiviral responses early in an infection. The two studies were recently published online in the journal Science, both of which were led by Jean-Laurent Casanova, an infectious disease geneticist at Rockefeller University. percent of women and 12.5
The tricuspid valve, one of the heart’s four valves, regulates blood flow from the right atrium to the right ventricle, preventing backflow between these chambers. In this blog, we discuss some of these new medical devices of 2024 that have improved patient outcomes and enhanced quality of life worldwide.
It is not uncommon for some in vitro diagnostic tests to suffer from poor positive predictive value, in part due to the rarity of the disease. Our readers will recall that this report was issued shortly after FDA’s release of its draft guidance documents seeking to regulate laboratory developed tests.
By Véronique Li, Senior Medical Device Regulation Expert & Jeffrey N. De Novos now play an important role in product advancement. Gibbs — In 1997, Congress wisely amended the Federal Food, Drug, and Cosmetic Act (FDCA) by adding Section 513(f)(2) to establish the De Novo process.
Complaints FDA discussed how LDT developers should handle the transition from the current Quality System Regulation (QSR)(21 CFR Part 820) to the recently promulgated Quality Management System Regulation (QMSR) that is scheduled to take effect on February 2, 2026. By Steven J. Gonzalez & Lisa M.
Eurofins Discovery will be conducting the IND-enabling in-vitro preclinical primary pharmacology and safety pharmacology studies on TD-0148A at its state-of-the-art facilities at Eurofins Cerep, DiscoverX and Panlabs. This approach makes Eurofins Discovery uniquely qualified to fulfill the needed studies for this innovative product.
Gibbs — It is widely expected that the fate of the VALID Act – and therefore the world of diagnostic regulation – will be determined in the next two weeks (see our previous post here ). Javitt & Jeffrey N. This is not a trivial matter. Telehealth has become increasing important to the health care system. Emphasis added).
The guidance for the voluntary STeP program provides additional support to developers of devices that significantly improve on existing products but do not meet all the criteria for the Breakthrough Devices Program. In vitro diagnostics that monitor and evaluate serious adverse events associated with newly approved drugs may also qualify.
Although the medical device industry is dedicated to advancing healthcare innovation, pursuing environmental responsibility has encountered obstacles mainly due to rigorous regulations. Coloplast’s product portfolio includes Ostomy Care, Continence Care, Wound and Skin Care, Interventional Urology and Voice and Respiratory Care.
It includes questions, text, logic, and prompts, and integrates databases such as FDA product codes and FDA-recognized voluntary consensus standards. It also presents specific questions to collect data from the submitter and provides links to relevant regulations and guidance documents.
Laboratories struggling to understand the myriad implications of being regulated as device “manufacturers” were hopeful that additional guidance would shed light on how to apply FDA’s existing medical device regulatory framework to their operations. 1] However, the focus of CLIA requirements is on laboratory processes, not specific assays.
Sartorius is a globally recognized player in the diagnostics industry, providing a variety of solutions for in vitro diagnostics kit (IVD) manufacturers. Sartorius understands the need for continuous innovation in the highly regulated and price-sensitive diagnostics market. Banczyk has expertise in medical and IVD filtration devices.
and global regulatory requirements for our oral treatment, PAXLOVID™, Pfizer undertakes in vitro work (e.g., and global regulators for all antiviral products and are carried out by many companies and academic institutions in the U.S. It is important to note that these studies are required by U.S. and around the world.
Before the FDA permits a pharmaceutical drug product to be lawfully marketed, sponsors are required to submit information about the product’s safety and efficacy so the FDA can determine : . The Food and Drug Administration (FDA) is the federal entity in the U.S. When Does an IND go into Effect? . The IND goes into effect; and .
. Product type: Nucleic acid testing (NAT) technologies that use real-time polymerase chain reaction (RT-PCR) for detection of SARS-CoV-2. Purpose of this notice : To ensure users of certain nucleic acid testing (NAT) technologies are aware of certain aspects of the instructions for use (IFU) for all products.
Baumhardt, Senior Medical Device Regulation Expert — On March 8th FDA granted Quidel’s Sofia 2 SARS Antigen+ FIA and Sofia 2 SARS Antigen+ FIA Control Swab Set. The authorization of this De Novo, with a formal classification of Class II, now opens the door for follow-up 510(k) submissions that declare this product as their predicate.
Nirmatrelvir has shown consistent in vitro antiviral activity against the following variants: Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1 I am grateful to have received four doses of the Pfizer-BioNTech vaccine and I am feeling well while experiencing very mild symptoms. FDA Emergency Use Authorization Statement. AUTHORIZED USE.
Mullen — FDA has been clearing over-the-counter (OTC) in vitro diagnostic (IVD) tests nearly since the beginning of its premarket regulation of devices. The first OTC IVD cleared by FDA was a qualitative dipstick urine glucose test in 1977, followed shortly thereafter by the first OTC pregnancy test clearance in 1978.
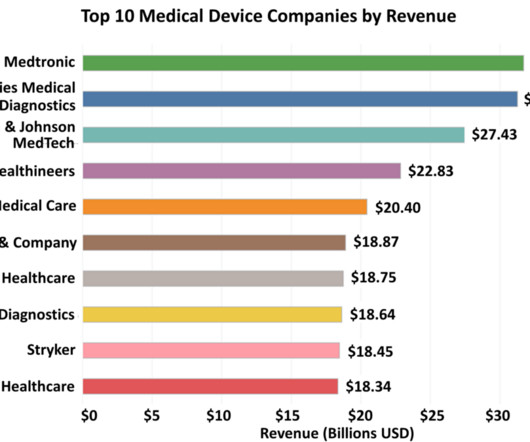
In Medtronic’s annual report, CEO Geoff Martha attributes their success in this area to their comprehensive suite of products and solutions, which equip clinicians with the tools necessary for optimal atrial fibrillation care. Note: When it comes to companies that report in foreign currencies, the conversion to U.S.
Etta Biotech”), to set up a high titer transient protein expression platform for high quality protein production using JS Bio’s transient transfection media. The two parties also plan to join force further to build a rapid protein production platform aiming at producing gram-scale protein for R&D, e. JS Bio’s parent company.
The test is planned for commercial launch as a CE-IVD ( in vitro diagnostic) certified product in the European Union in Q1 2021. “Applying 3a’s proprietary enhanced RNA technology to PCR testing was a logical next step in our product development pipeline,” said Dr. Heinrich Jehle, managing director of 3a-Diagnostics.
In vitro diagnostic (IVD) devices are tests used on human biospecimens (e.g., Additionally, while the Food and Drug Administration (FDA) regulates all IVDs as medical devices, the agency has generally not enforced device regulations for certain IVDs designed, manufactured, and used within a single CLIA-certified laboratory.
We organize all of the trending information in your field so you don't have to. Join 21,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content